Другие названия и синонимы
Metachromatic leukodystrophy.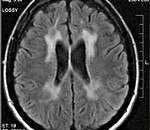
МКБ-10 коды
- МКБ-10
- E75.2 Другие сфинголипидозы
|
|
Описание
Метахроматическая лейкодистрофия. Аутосомно - рецессивно наследуемая демиелинизирующая патология ЦНС, отличительной особенностью которой является метахроматическое окрашивание зон демиелинизации. В клинике преобладает задержка развития, парезы, судорожные приступы, экстрапирамидные и мозжечковые расстройства, нарушение зрения. В ходе диагностики проводится исследование цереброспинальной жидкости, уровня сульфатидов, активности арилсульфатазы А, КТ/МРТ головного мозга, генетические обследования. Возможна пренатальная диагностика. Лечение симптоматическое, в отдельных случаях проводится трансплантация пуповинной крови или стволовых клеток.
Дополнительные факты
Метахроматическая лейкодистрофия - один из основных видов лейкодистрофии, характеризующийся, как и другие ее типы, задержкой психомоторного развития, расстройством психики, спастическими парезами, мозжечковым синдромом, экстрапирамидными нарушениями, судорожным синдромом и атрофией зрительных нервов. Встречается в популяции с частотой 1 случай на 40 тыс. Наследуется аутосомно-рецессивно. Впервые выделена как нозологическая форма в 1910 г. Альцгеймером, подробно описана в 1925 г. Шольцем. Время манифестации заболевания существенно варьирует и обуславливает его клиническое течение. В соответствии с этими особенностями выделяют несколько вариантов патологии: врожденную (раннедетскую) форму, позднедетскую форму Гринфилда, ювенильную форму Шольца и взрослую форму. Наиболее часто наблюдается познедетская форма. В виду отсутствия эффективных способов лечения метахроматическая лейкодистрофия является проблемной патологией для современной педиатрии, неврологии и генетики.
Причины
Основным субстратом заболевания выступает дефект расположенного на 22-й хромосоме гена, ответственного за синтез арилсульфатазы А. Результатом дефицита указанного энзима является блокировка метаболизма сульфатида в галактоцереброзид. Сульфатиды откладываются в белом веществе головного и спинного мозга, коже, соматических органах (печени, легких, сердце, почках) и в костях. Однако при этом функция внутренних органов не страдает (исключением является желчный пузырь), а в мозговом веществе происходят прогрессирующие дегенеративные изменения, ведущие к гибели заболевшего.
Отложение сульфатидов в нервной ткани приводит к разрушению миелина и накоплению продуктов его распада. В процесс демиелинизации вовлекаются не только структуры ЦНС, но и периферические нервные стволы. В головном мозге формируются диффузные очаги демиелинизации, дающие при их гистохимическом исследовании метахроматическое окрашивание.
Отложение сульфатидов в нервной ткани приводит к разрушению миелина и накоплению продуктов его распада. В процесс демиелинизации вовлекаются не только структуры ЦНС, но и периферические нервные стволы. В головном мозге формируются диффузные очаги демиелинизации, дающие при их гистохимическом исследовании метахроматическое окрашивание.
Клиническая картина
Врожденная метахроматическая лейкодистрофия дебютирует до 3-месячного возраста задержкой развития и эписиндромом. Быстро присоединяется спастический парез, расстройства глотания. Дети погибают на 1-ом году жизни.
Позднедетская метахроматическая лейкодистрофия (поздняя инфантильная форма) обычно манифестирует на 2-ом году жизни ухудшением моторики и задержкой психического развития. Отмечается неустойчивая походка, понижение мышечного тонуса. В течении позднедетской формы различают 4 периода. Первый характеризуется мышечной гипотонией и слабостью, угасанием сухожильных рефлексов, затруднением ходьбы. Длится от 1-3 мес. до 1 года, иногда и дольше. Второй период знаменуется выраженным отставанием в интеллектуальной сфере, сменой гипотонии на гипертонию мышц. Ребенок утрачивает способность стоять. Наблюдается дизартрия, атрофия зрительных нервов. Период занимает несколько месяцев. В третьем периоде отмечается тетраплегия, тяжелая олигофрения, бульбарный и псевдобульбарный паралич. Четвертый период сопровождается утратой реакций на окружающие события. Ребенок не говорит, не может принимать пищу, развивается полная слепота. При хорошем уходе и кормлении через гастростому или желудочный зонд данный период может продолжаться 1-2 года.
Ювенильная метахроматическая лейкодистрофия начинается после 3-летнего возраста в периоде до 10 лет (чаще в возрасте около 6 лет). Дебютирует эмоциональной лабильностью, изменениями в поведении, снижением когнитивных функций (у школьников происходит резкое ухудшение успеваемости), атаксией, затруднениями при ходьбе. Зачастую отмечаются эпиприступы. Заболевание неуклонно прогрессирует и, в зависимости от начала симптоматики, к 10-15 годам жизни приводит к летальному исходу.
Позднедетская метахроматическая лейкодистрофия (поздняя инфантильная форма) обычно манифестирует на 2-ом году жизни ухудшением моторики и задержкой психического развития. Отмечается неустойчивая походка, понижение мышечного тонуса. В течении позднедетской формы различают 4 периода. Первый характеризуется мышечной гипотонией и слабостью, угасанием сухожильных рефлексов, затруднением ходьбы. Длится от 1-3 мес. до 1 года, иногда и дольше. Второй период знаменуется выраженным отставанием в интеллектуальной сфере, сменой гипотонии на гипертонию мышц. Ребенок утрачивает способность стоять. Наблюдается дизартрия, атрофия зрительных нервов. Период занимает несколько месяцев. В третьем периоде отмечается тетраплегия, тяжелая олигофрения, бульбарный и псевдобульбарный паралич. Четвертый период сопровождается утратой реакций на окружающие события. Ребенок не говорит, не может принимать пищу, развивается полная слепота. При хорошем уходе и кормлении через гастростому или желудочный зонд данный период может продолжаться 1-2 года.
Ювенильная метахроматическая лейкодистрофия начинается после 3-летнего возраста в периоде до 10 лет (чаще в возрасте около 6 лет). Дебютирует эмоциональной лабильностью, изменениями в поведении, снижением когнитивных функций (у школьников происходит резкое ухудшение успеваемости), атаксией, затруднениями при ходьбе. Зачастую отмечаются эпиприступы. Заболевание неуклонно прогрессирует и, в зависимости от начала симптоматики, к 10-15 годам жизни приводит к летальному исходу.
Диагностика
Диагностические мероприятия имеют комплексный характер и позволяют отдифференцировать метахроматическую лейкодистрофию от других нейродегенеративных заболеваний и других форм лейкодистрофии. Проводится люмбальная пункция, при исследовании цереброспинальной жидкости определяется повышенная концентрация белка, дефицит арилсульфатазы, белково-клеточная диссоциация. Электромиография выявляет увеличение длительности проведения нервных импульсов. КТ головного мозга визуализирует расширенные желудочки и определяет пониженную плотность белого церебрального вещества. МРТ головного мозга выявляет очаги демиелинизации.
Важное диагностическое значение имеют биохимические исследования: определение сульфатидов в крови и моче, оценка активности арилсульфатазы А в лейкоцитах взятой на анализ крови. Пациентам показана консультация генетика с проведением генеалогического исследования; возможна ДНК-диагностика. Разработан также метод пренатальной диагностики патологии - определение активности арилсульфатазы А в клетках полученной при амниоцентезе амниотической жидкости. Исследования показали, что у носителей патогенного гена также отмечается сниженный уровень арилсульфатазы А. В связи с этим вместо пренатальной диагностики плода предлагается обследовать родителей будущего ребенка, чтобы избежать медаборта плода, являющегося лишь носителем дефектного гена.
Важное диагностическое значение имеют биохимические исследования: определение сульфатидов в крови и моче, оценка активности арилсульфатазы А в лейкоцитах взятой на анализ крови. Пациентам показана консультация генетика с проведением генеалогического исследования; возможна ДНК-диагностика. Разработан также метод пренатальной диагностики патологии - определение активности арилсульфатазы А в клетках полученной при амниоцентезе амниотической жидкости. Исследования показали, что у носителей патогенного гена также отмечается сниженный уровень арилсульфатазы А. В связи с этим вместо пренатальной диагностики плода предлагается обследовать родителей будущего ребенка, чтобы избежать медаборта плода, являющегося лишь носителем дефектного гена.
Лечение
Терапия, способная предотвратить летальный исход заболевания, пока не найдена. Осуществляется лечение, направленное на купирование его отдельных симптомов: противосудорожная терапия, борьба с контрактурами суставов; на поздних стадиях - профилактика пролежней, парентеральное питание. Проводится поиск более эффективных способов лечения, особенно в области генной терапии.
Предпринимаются попытки лечения путем трансплантации костного мозга и пуповинной крови. У ряда пациентов пересадка стволовых клеток позволяет добиться временной стабилизации состояния и замедления прогрессирования патологических изменений. Донором стволовых клеток обычно выступает родственник больного, не имеющий лейкодистрофии и тщательно обследованный. Однако следует помнить, что сама трансплантация может повлечь за собой целый ряд осложнений: отторжение, интеркурентные инфекции, реакцию «трансплантат против хозяина». Кроме того, при быстром прогрессировании симптомов летальный исход опережает наступление эффекта трансплантации.
Предпринимаются попытки лечения путем трансплантации костного мозга и пуповинной крови. У ряда пациентов пересадка стволовых клеток позволяет добиться временной стабилизации состояния и замедления прогрессирования патологических изменений. Донором стволовых клеток обычно выступает родственник больного, не имеющий лейкодистрофии и тщательно обследованный. Однако следует помнить, что сама трансплантация может повлечь за собой целый ряд осложнений: отторжение, интеркурентные инфекции, реакцию «трансплантат против хозяина». Кроме того, при быстром прогрессировании симптомов летальный исход опережает наступление эффекта трансплантации.
Прогноз
Пока не найдена эффективная терапия лейкодистрофий, она имеет крайне неблагоприятный прогноз. При врожденной форме дети не доживают до 1 года. При позднедетском варианте продолжительность болезни, как правило, не превышает 4 года, при ювенильном - 6 лет. Среди пациентов со взрослой формой известны отдельные случаи, когда продолжительность жизни составила около 50 лет. Пока единственным способом предупреждения лейкодистрофии является недопущение рождения ребенка, имеющего соответствующую генную мутацию. В этом плане основное значение имеет генетическое консультирование пар, планирующих деторождение, а также обследование будущих родителей из группы риска на степень активности арилсульфатазы А.
|
|