ICD-10 codes
- ICD-10
- E75.2 Other sphingolipidosis
Description
Лейкодистрофия. Нейродегенеративное заболевание, обусловленное наследственным нарушением обмена веществ с накоплением в головном и спинном мозге метаболитов, провоцирующих разрушение миелина. Манифестирует в основном в детском возрасте задержкой психомоторного развития, двигательными расстройствами, поражением зрительных и слуховых нервов, гидроцефалией, эпилептическими приступами. Диагностируется лейкодистрофия по данным неврологического статуса, анамнеза, генетических исследований, МРТ или КТ картины головного мозга, биохимических анализов. Лечение симптоматическое. При раннем выявлении и медленном прогрессировании возможна трансплантация пуповинной крови или костного мозга.
Additional facts
Лейкодистрофия получила свое название в связи с поражением белого вещества мозга (с греческого leukos - белый). Различают около 60 разновидностей лейкодистрофии, определяющихся видом генной аномалии и возрастом манифестации клинических проявлений. Наряду с отдельными воспалительными поражениями ЦНС (например, лейкоэнцефалитом Шильдера) лейкодистрофия относится к синдрому диффузного склероза мозга. При этом доминирующее поражение миелина сближает ее с демиелинизирующими заболеваниями (рассеянным склерозом, РЭМ и пр. ), а отдельные формы можно отнести к липидозам.
К основным формам лейкодистрофии относятся метахроматическая, суданофильная, глобоидно-клеточная, дегенерация Ван-Богарта-Бертрана, болезнь Александера, вариант Галлервордена-Шпатца. Наиболее распространены первые 3 вида лейкодистрофии. Их встречаемость колеблется от 0,4 до 1 случая на 100 тыс. новорожденных. Ряд форм лейкодистрофии являются настолько редкими, что в мировой литературе по неврологии описано всего несколько сотен их клинических наблюдений. В зависимости от возрастного периода, в котором дебютирует лейкодистрофия, каждая ее форма может подразделяться на инфантильный, поздний инфантильный, ювенильный и взрослый вариант.
К основным формам лейкодистрофии относятся метахроматическая, суданофильная, глобоидно-клеточная, дегенерация Ван-Богарта-Бертрана, болезнь Александера, вариант Галлервордена-Шпатца. Наиболее распространены первые 3 вида лейкодистрофии. Их встречаемость колеблется от 0,4 до 1 случая на 100 тыс. новорожденных. Ряд форм лейкодистрофии являются настолько редкими, что в мировой литературе по неврологии описано всего несколько сотен их клинических наблюдений. В зависимости от возрастного периода, в котором дебютирует лейкодистрофия, каждая ее форма может подразделяться на инфантильный, поздний инфантильный, ювенильный и взрослый вариант.
Reasons
В своей основе каждая лейкодистрофия имеет генетическую аномалию определенного фермента. Вид аномалии и локализация генной мутации пока установлены лишь для наиболее встречающихся форм патологии. В большинстве случаев лейкодистрофия имеет аутосомно-рецессивный путь наследственной передачи, однако отдельные ее формы могут наследоваться сцеплено с полом. Кроме того, не одиноки случаи спонтанных мутаций. Генетически детерминированный энзимный дефект ведет к обменным нарушениям (чаще в метаболизме липидов) с отложением определенного метаболита в нервных структурах и отдельных соматических органах, в первую очередь в печени и почках.
Следствием метаболической аномалии является разрушение миелина оболочек нервных стволов и проводящих путей, гибель нейронов с замещением их разрастающейся глиальной тканью. Морфологически лейкодистрофия характеризуется диффузными и симметрично расположенными в полушариях головного мозга зонами гибели миелина, скоплением продуктов миелинового распада, усиленной пролиферацией глии. В отдельных нозологических вариантах лейкодистрофия имеет специфическую морфологическую картину - метахроматическое или суданофильное окрашивание продуктов миелинового распада, скопление в зонах демиелинизации глобоидных клеток.
Следствием метаболической аномалии является разрушение миелина оболочек нервных стволов и проводящих путей, гибель нейронов с замещением их разрастающейся глиальной тканью. Морфологически лейкодистрофия характеризуется диффузными и симметрично расположенными в полушариях головного мозга зонами гибели миелина, скоплением продуктов миелинового распада, усиленной пролиферацией глии. В отдельных нозологических вариантах лейкодистрофия имеет специфическую морфологическую картину - метахроматическое или суданофильное окрашивание продуктов миелинового распада, скопление в зонах демиелинизации глобоидных клеток.
Symptoms
В большинстве случаев лейкодистрофия дебютирует в раннем детском возрасте. Новорожденные, как правило, выглядят здоровыми. Определенный период они нормально развиваются, а затем постепенно возникают различные неврологические симптомы, отличающиеся неуклонным прогрессированием. Скорость нарастания симптомов тем выше, чем раньше манифестировала лейкодистрофия. Ведущими проявлениями выступают прогрессирующая олигофрения, ухудшение зрения, тугоухость, эписиндром, спастические парезы. Первыми симптомами лейкодистрофии могут быть атаксия, мышечно-тонические расстройства (гипо- или гипертонус, мышечные подергивания), экстрапирамидные проявления, изменения поведения. Затем возникают эпиприступы, бульбарные проявления, снижается слух и зрение, отмечается интеллектуальное снижение с постепенной утратой ранее приобретенных навыков. Сенсорные расстройства не характерны. На поздних этапах развития болезни наблюдаются параличи, выраженная олигофрения, грубое расстройство глотания, амавроз, глухота. В терминальной фазе обычно отмечается децеребрационная ригидность.
Ассоциированные симптомы: Слабость мышц (парез). Судороги. Тетрапарез. Тонико-клонические судороги. Тремор.
Ассоциированные симптомы: Слабость мышц (парез). Судороги. Тетрапарез. Тонико-клонические судороги. Тремор.
Classification
Метахроматическая лейкодистрофия в зависимости от манифестации имеет 4 варианта. Врожденный вариант дебютирует в первые 1-3 мес. жизни задержкой развития и судорожным синдромом; дети не достигают возраста 1 года. Позднедетский вариант метахроматической лейкодистрофии начинается в период от 1 до 3 лет с мышечной гипотонии и слабости, атаксии, задержки психического развития (ЗПР). Затем формируется спастическая тетраплегия, афазия, псевдобульбарный синдром. В редких случаях пациенты доживают до 10-летнего возраста. Ювенильный вариант манифестирует в 4-6 лет и длится в среднем 7 лет. Взрослый вариант дебютирует в третьей декаде жизни, иногда позднее, продолжительность жизни пациентов от начала клиники варьирует в пределах 10-20 лет.
Суданофильная лейкодистрофия наследуется сцеплено с Х-хромосомой и имеет несколько разновидностей. Лейкодистрофия Пелицеуса-Мерцбахера может стартовать на 1-ом году жизни или в 3-4 года. Первым признаком является крупноразмашистый нистагм, позже возникает ЗПР, мозжечковая атаксия, гиперкинезы, парезы. Наибольшее прогрессирование происходит в возрасте до 10 лет, затем заболевание принимает замедленное течение с длительными ремиссиями. Пациенты могут жить до зрелого возраста. Адренолейкодистрофия - вариант, при котором лейкодистрофия сочетается с надпочечниковой недостаточностью. Характеризуется прогрессирующим течением с летальным исходом спустя 6-8 лет от начала клиники.
Глобоидно. Клеточная лейкодистрофия (болезнь Краббе) - липоидоз с накоплением в очагах демиелинизации галактоцереброзида и образованием больших округлых глобоидных клеток. Раннедетский вариант развивается в первом полугодии жизни с гипервозбудимости и периодической гипертермии, задерживается психомоторное развитие, нарастает тонус мышц, затем развивается спастический тетрапарез, олигофрения, эписиндром, возможен опистотонус. В годовалом возрасте наступает летальный исход. Позднедетский вариант более редкий, манифестирует ухудшением зрения.
Спонгиозная дегенерация Ван. Богарта - Бертрана характеризуется эписиндромом, гиперсомнией, выраженной гидроцефалией с увеличением размеров головы, вызывающей амавроз атрофией зрительных нервов. Резкая внутричерепная гипертензия приводит к расхождению черепных швов, регистрируемому при рентгенографии черепа. Пациенты с этой формой лейкодистрофии погибают до 3-летнего возраста.
Болезнь Александера (лейкодистрофия с волокнистой формацией) обусловлена мутацией гена, ответственного за синтез GFAP белка. В результате происходит накопление в клетках глии аномального GFAP белка, содержащего волокна Розенталя. Неонатальный вариант имеет тяжелое течение с летальным исходом к концу 1-го года. Инфантильный вариант встречается примерно в половине случаев, проявляется в первые 1-2 года жизни ЗПР, затем присоединяются спастические парезы, атаксия, гидроцефалия. Дети погибают спустя несколько лет. Ювенильная лейкодистрофия Александера дебютирует в период от 4-х до 10-летнего возраста, протекает с преимущественно стволовой симптоматикой. Продолжительность жизни колеблется в пределах 10-30 лет. Взрослый вариант отличается поздней манифестацией и относительно медленным течением в пределах 10 и более лет.
Суданофильная лейкодистрофия наследуется сцеплено с Х-хромосомой и имеет несколько разновидностей. Лейкодистрофия Пелицеуса-Мерцбахера может стартовать на 1-ом году жизни или в 3-4 года. Первым признаком является крупноразмашистый нистагм, позже возникает ЗПР, мозжечковая атаксия, гиперкинезы, парезы. Наибольшее прогрессирование происходит в возрасте до 10 лет, затем заболевание принимает замедленное течение с длительными ремиссиями. Пациенты могут жить до зрелого возраста. Адренолейкодистрофия - вариант, при котором лейкодистрофия сочетается с надпочечниковой недостаточностью. Характеризуется прогрессирующим течением с летальным исходом спустя 6-8 лет от начала клиники.
Глобоидно. Клеточная лейкодистрофия (болезнь Краббе) - липоидоз с накоплением в очагах демиелинизации галактоцереброзида и образованием больших округлых глобоидных клеток. Раннедетский вариант развивается в первом полугодии жизни с гипервозбудимости и периодической гипертермии, задерживается психомоторное развитие, нарастает тонус мышц, затем развивается спастический тетрапарез, олигофрения, эписиндром, возможен опистотонус. В годовалом возрасте наступает летальный исход. Позднедетский вариант более редкий, манифестирует ухудшением зрения.
Спонгиозная дегенерация Ван. Богарта - Бертрана характеризуется эписиндромом, гиперсомнией, выраженной гидроцефалией с увеличением размеров головы, вызывающей амавроз атрофией зрительных нервов. Резкая внутричерепная гипертензия приводит к расхождению черепных швов, регистрируемому при рентгенографии черепа. Пациенты с этой формой лейкодистрофии погибают до 3-летнего возраста.
Болезнь Александера (лейкодистрофия с волокнистой формацией) обусловлена мутацией гена, ответственного за синтез GFAP белка. В результате происходит накопление в клетках глии аномального GFAP белка, содержащего волокна Розенталя. Неонатальный вариант имеет тяжелое течение с летальным исходом к концу 1-го года. Инфантильный вариант встречается примерно в половине случаев, проявляется в первые 1-2 года жизни ЗПР, затем присоединяются спастические парезы, атаксия, гидроцефалия. Дети погибают спустя несколько лет. Ювенильная лейкодистрофия Александера дебютирует в период от 4-х до 10-летнего возраста, протекает с преимущественно стволовой симптоматикой. Продолжительность жизни колеблется в пределах 10-30 лет. Взрослый вариант отличается поздней манифестацией и относительно медленным течением в пределах 10 и более лет.
Diagnostics
Диагностический поиск требует привлечения ряда специалистов: невролога, педиатра, медицинского генетика, для диагностики расстройств зрения и слуха - отоларинголога и офтальмолога. Важное значение имеет изучение анамнеза болезни (возраст и симптомы дебюта, последовательность развития клиники) и семейного анамнеза (наличие лейкодистрофии у родственников). Нейросонография через родничок и эхо-энцефалография у пациентов более старшего возраста, как правило, выявляет повышение интракраниального давления. Лейкодистрофия сопровождается существенным увеличением концентрации белка, обусловленным разрушением церебральных клеток, что определяется при исследовании цереброспинальной жидкости.
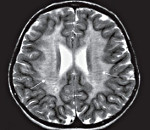
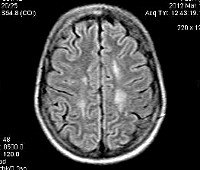
С целью диагностики вида метаболической аномалии проводится целый ряд биохимических тестов с определением уровня ферментов и накапливающихся метаболитов. Очаги демиелинизации хорошо визуализируются при помощи МРТ, могут быть обнаружены и на КТ головного мозга. Обычно демиелинизация видна на МРТ головного мозга еще до клинической манифестации лейкодистрофии. Благодаря развитию генетики, лейкодистрофия имеет разработанную ДНК-диагностику, а отдельные ее формы (метахроматическая, адренолейкодистрофия, глобоидно-клеточная) - возможность пренатального диагностирования.
С целью диагностики вида метаболической аномалии проводится целый ряд биохимических тестов с определением уровня ферментов и накапливающихся метаболитов. Очаги демиелинизации хорошо визуализируются при помощи МРТ, могут быть обнаружены и на КТ головного мозга. Обычно демиелинизация видна на МРТ головного мозга еще до клинической манифестации лейкодистрофии. Благодаря развитию генетики, лейкодистрофия имеет разработанную ДНК-диагностику, а отдельные ее формы (метахроматическая, адренолейкодистрофия, глобоидно-клеточная) - возможность пренатального диагностирования.
Treatment
На сегодняшний день лейкодистрофия не имеет эффективных способов терапии, позволяющих купировать прогрессирование симптомов. Проводится симптоматическое лечение - в основном дегидратационная и антиконвульсантная терапия. Единственным методом, способным увеличить продолжительность жизни пациентов с лейкодистрофией и улучшить качество их жизни, является трансплантация пуповинной крови или пересадка костного мозга. Трансплантация приводит к нормализации метаболизма. Однако этот процесс занимает длительное время (от 12 до 24 мес. ), в течение которого продолжается прогрессирование лейкодистрофии. Поэтому зачастую тяжелая инвалидизация или гибель пациента наступает даже после успешной трансплантации.
Следует подчеркнуть, что трансплантация никак не влияет на уже развившийся неврологический дефицит, она лишь позволяет приостановить его дальнейшее прогрессирование. В связи с тем, что эффект такого лечения наступает спустя 1-2 года, оно целесообразно в случае ранней доклинической диагностики лейкодистрофии (при соответствующей настороженности родителей рожденного ребенка в связи с наличием подобной патологии в семье) или при медленно прогрессирующем варианте течения. Кроме того, необходимо учитывать, что трансплантация связана с риском ряда серьезных осложнений, таких как отторжение, реакция «трансплантат против хозяина», развитие инфекций.
Следует подчеркнуть, что трансплантация никак не влияет на уже развившийся неврологический дефицит, она лишь позволяет приостановить его дальнейшее прогрессирование. В связи с тем, что эффект такого лечения наступает спустя 1-2 года, оно целесообразно в случае ранней доклинической диагностики лейкодистрофии (при соответствующей настороженности родителей рожденного ребенка в связи с наличием подобной патологии в семье) или при медленно прогрессирующем варианте течения. Кроме того, необходимо учитывать, что трансплантация связана с риском ряда серьезных осложнений, таких как отторжение, реакция «трансплантат против хозяина», развитие инфекций.