Другие названия и синонимы
Progressive Erba-Roth muscular dystrophy.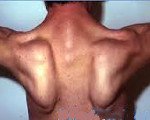
МКБ-10 коды
- МКБ-10
- G71.0 Мышечная дистрофия
|
|
Описание
Прогрессирующая мышечная дистрофия Эрба. Рота - аутосомно - рецессивная наследственная миодистрофия, отличающаяся полиморфизмом клинических проявлений и вариативностью скорости прогрессирования симптомов. Может носить нисходящий характер, начинаться со слабости в проксимальных отделах рук, но чаще имеет стандартный восходящий тип распространения мышечных изменений. Диагностика осуществляется методами неврологического осмотра, электрофизиологического тестирования, генеалогического исследования и генетического анализа, по показаниям проводится гистологический анализ биоптата мышечной ткани. Лечение только симптоматическое, позволяющее лишь продлить двигательную активность пациентов. Итог заболевания - полная обездвиженность.
Дополнительные факты
Прогрессирующая мышечная дистрофия Эрба-Рота - самый полиморфный вариант наследственной миодистрофии. От других видов прогрессирующей мышечной дистрофии вариант Эрба-Рота отличается вариабельностью как времени дебюта заболевания, так его клинической картины и течения. Заболевание описано в 1882 г. немецким неврологом Вильгельмом Эрбом. Одновременно в России изучением этой патологии занимался В. Рот, для обозначения миодистрофии он ввел термин «мышечная сухотка». В современной мировой и отечественной неврологии в честь этих исследователей употребляется название «прогрессирующая мышечная дистрофия Эрба-Рота». Частота встречаемости заболевания находится в пределах от 1,5 до 2,5 случаев на 100 тыс. населения.
Причины
Субстратом дистрофии Эрба-Рота являются патологические метаболические и структурные изменения в мышечной ткани (миопатия). Они возникают в результате генетических мутаций, приводящих к недостатку или полному прекращению синтеза белков, являющихся необходимыми структурными компонентами миоцитов. На сегодняшний день генетике известно не менее 9 хромосомных локусов, аберрации в которых приводят к развитию миодистрофии Эрба-Рота. Чаще всего наблюдаются мутации в локусах 15q15-q21. 1, 13q.
Около 30% генных аномалий возникают de novo, остальные носят семейный характер. Наследуется мышечная дистрофия Эрба-Рота аутосомно-рецессивно. Болеют как мальчики, так и девочки. Патология проявляется, если ребенок получает аномальный ген от каждого из родителей. В случае, когда оба родителя выступают носителями аберрантного гена, но сами не болеют, вероятность развития миодистрофии у ребенка составляет 25%.
Около 30% генных аномалий возникают de novo, остальные носят семейный характер. Наследуется мышечная дистрофия Эрба-Рота аутосомно-рецессивно. Болеют как мальчики, так и девочки. Патология проявляется, если ребенок получает аномальный ген от каждого из родителей. В случае, когда оба родителя выступают носителями аберрантного гена, но сами не болеют, вероятность развития миодистрофии у ребенка составляет 25%.
Клиническая картина
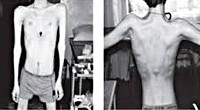
Прогрессирующая мышечная дистрофия Эрба-Рота манифестирует в среднем в возрасте 13-16 лет. Однако известны отдельные случаи дебюта болезни в раннем детском возрасте и в возрасте после 20 лет. Мышечная слабость и атрофии возникают в первую очередь в мускулатуре тазового пояса и проксимальных отделов ног. Отмечаются затруднения ходьбы по лестнице, подъема из положения присев на корточки. Типичен симптом Говерса - если пациент сидел на полу, то для того, чтобы подняться, он использует собственное тело как опору.
Со временем дистрофические изменения распространяются на мышечные группы туловища и рук. Такой тип распространения миодистрофии носит название восходящий. Он наиболее типичен для большинства наследственных мышечных дистрофий. Однако в некоторых случаях дистрофии Эрба-Рота наблюдается нисходящий тип распространения патологического процесса, когда мышечная слабость возникает вначале в руках, затем в тазовом поясе, а через несколько лет в мышцах ног.
Тотальная гипотрофия мышц туловища приводит к тому, что у пациентов начинают выступать лопатки (т. н. «крыловидные лопатки»), талия становиться очень тонкой (т. н. «осиная талия»), усиливается поясничный лордоз, живот выпячивается вперед. Характерен симптом свободных надплечий - при попытке приподнять больного вверх, удерживая его за подмышки, плечи пациента свободно движутся вверх и голова будто бы «проваливается» между ними. Поражение лицевых мышц влечет за собой гипомимию (т. н. «лицо сфинкса»), неполное закрытие век, выворачивание и утолщение губ.
Ассоциированные симптомы: Запор. Слабость в ногах. Слабость в руках. Слабость мышц (парез). Слабость мышц лица. Утиная походка.
Со временем дистрофические изменения распространяются на мышечные группы туловища и рук. Такой тип распространения миодистрофии носит название восходящий. Он наиболее типичен для большинства наследственных мышечных дистрофий. Однако в некоторых случаях дистрофии Эрба-Рота наблюдается нисходящий тип распространения патологического процесса, когда мышечная слабость возникает вначале в руках, затем в тазовом поясе, а через несколько лет в мышцах ног.
Тотальная гипотрофия мышц туловища приводит к тому, что у пациентов начинают выступать лопатки (т. н. «крыловидные лопатки»), талия становиться очень тонкой (т. н. «осиная талия»), усиливается поясничный лордоз, живот выпячивается вперед. Характерен симптом свободных надплечий - при попытке приподнять больного вверх, удерживая его за подмышки, плечи пациента свободно движутся вверх и голова будто бы «проваливается» между ними. Поражение лицевых мышц влечет за собой гипомимию (т. н. «лицо сфинкса»), неполное закрытие век, выворачивание и утолщение губ.
Ассоциированные симптомы: Запор. Слабость в ногах. Слабость в руках. Слабость мышц (парез). Слабость мышц лица. Утиная походка.
Диагностика
Миодистрофия Эрба-Рота диагностируется неврологом и генетиком при сопоставлении данных анамнеза (возраст дебюта заболевания, последовательность развития симптомов), неврологического статуса пациента, ЭФИ нервно-мышечной системы, генеалогических данных, результатов анализа ДНК и микроскопического исследования мышечной ткани.
Дифференциальная диагностика
Дифференцировать миодистрофию Эрба-Рота приходится от других форм этого заболевания (прогрессирующей дистрофии Дюшенна, миодистрофии Дрейфуса и Беккера), дерматомиозита, полимиозита, бокового амиотрофического склероза, токсической миопатии и тд.
При неврологическом осмотре обращает на себя внимание снижение мышечной силы в мускулатуре проксимальных отделов ног и рук, гипотония и гипотрофия указанных мышц, гипорефлексия или полное выпадение локтевых и коленных рефлексов, сохранность всех видов чувствительности. ЭМГ и ЭНГ свидетельствуют о первичном поражении мышечной ткани при сохранности проведения импульсов по нервным стволам.
Генеалогическое исследование подтверждает аутосомно-рецессивный характер наследования. Исследование ДНК может выявить наличие генных мутаций. Однако отрицательный результат исследования не опровергает диагноз, поскольку не всякая мутация может быть обнаружена. Отрицательный анализ ДНК является показанием к биопсии мышц. В биоптате обнаруживаются различные по толщине мышечные волокна, уменьшенное число мышечных ядер, некротические и склеротические изменения.
Резкое повышение уровня креатинфосфокиназы характерно для начального периода дистрофии Эрба-Рота, затем происходит постепенное понижение этого показателя вплоть до нормальных цифр. Обзорная рентгенография грудной клетки позволяет выявить расширение границ сердца, наличие воспалительных изменений легочной ткани. ЭКГ зачастую определяет аритмию и нарушения проводимости. При помощи УЗИ сердца можно диагностировать кардиомиопатию. Для оценки степени сердечных нарушений требуется консультация кардиолога, при подозрении на пневмонию - консультация пульмонолога.
При неврологическом осмотре обращает на себя внимание снижение мышечной силы в мускулатуре проксимальных отделов ног и рук, гипотония и гипотрофия указанных мышц, гипорефлексия или полное выпадение локтевых и коленных рефлексов, сохранность всех видов чувствительности. ЭМГ и ЭНГ свидетельствуют о первичном поражении мышечной ткани при сохранности проведения импульсов по нервным стволам.
Генеалогическое исследование подтверждает аутосомно-рецессивный характер наследования. Исследование ДНК может выявить наличие генных мутаций. Однако отрицательный результат исследования не опровергает диагноз, поскольку не всякая мутация может быть обнаружена. Отрицательный анализ ДНК является показанием к биопсии мышц. В биоптате обнаруживаются различные по толщине мышечные волокна, уменьшенное число мышечных ядер, некротические и склеротические изменения.
Резкое повышение уровня креатинфосфокиназы характерно для начального периода дистрофии Эрба-Рота, затем происходит постепенное понижение этого показателя вплоть до нормальных цифр. Обзорная рентгенография грудной клетки позволяет выявить расширение границ сердца, наличие воспалительных изменений легочной ткани. ЭКГ зачастую определяет аритмию и нарушения проводимости. При помощи УЗИ сердца можно диагностировать кардиомиопатию. Для оценки степени сердечных нарушений требуется консультация кардиолога, при подозрении на пневмонию - консультация пульмонолога.
Лечение
Этиопатогенетическая терапия пока не разработана. Симптоматическое лечение направлено на как можно более длительное сохранение двигательной способности пациента. С этой целью применяют медикаментозные курсы, включающие АТФ, витамины Е и группы В, тиоктовую кислоту и тд Занятия лечебной физкультурой должны проводиться ежедневно и включать упражнения на все группы мышц. Регулярно назначаются курсы массажа и физиопроцедуры.
При поражении сердечной мышцы рекомендован инозин, сердечные гликозиды, антиаритмики. При развитии контрактур может потребоваться ортопедическое лечение. Выраженное снижение жизненной емкости легких из-за атрофии дыхательных мышц служит показанием к ИВЛ.
При поражении сердечной мышцы рекомендован инозин, сердечные гликозиды, антиаритмики. При развитии контрактур может потребоваться ортопедическое лечение. Выраженное снижение жизненной емкости легких из-за атрофии дыхательных мышц служит показанием к ИВЛ.
|
|