Другие названия и синонимы
Distal motor neuropathy.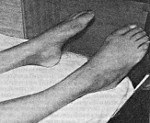
МКБ-10 коды
- МКБ-10
- G62 Другие полинейропатии
|
|
Описание
Дистальная моторная нейропатия. Генетически гетерогенная группа наследственных заболеваний, которые объединяет нарушение иннервации мышц конечностей с развитием атрофии без патологии чувствительных волокон. Симптомами этих состояний являются медленно прогрессирующая слабость мышц ног, рук и плечевого пояса, а также уменьшение мышечной массы в указанных областях. Иногда может выявляться нарушение вибрационной чувствительности. Диагностика дистальной моторной нейропатии осуществляется на основании данных осмотра пациента, изучения его анамнеза, электромиографии и молекулярно-генетических исследований. В некоторых случаях производят гистологическое изучение мышечной ткани. Эффективного лечения данной патологии не существует, для замедления прогрессирования заболевания используют витаминотерапию и препараты, улучшающие трофику нервных тканей.
Дополнительные факты
Дистальная моторная нейропатия (дистальная спинальная амиотрофия) - группа генетически обусловленных патологий различной природы, характеризующихся нарушением двигательной иннервации мышц конечностей с развитием атрофии. Несмотря на огромное количество разновидностей, встречаемость таких состояний довольно низка - в зависимости от типа ее значение колеблется в пределах 1-40:1000000. Некоторые формы дистальной моторной нейропатии известны с давнего времени. В настоящее время достижения современной генетики позволили дифференцировать ряд заболеваний, которые обладают одинаковыми фенотипическими проявлениями, но имеют разную генетическую подоплеку. По мнению многих исследователей, изучение данного состояния пока не завершено - некоторые гены не идентифицированы, возможно, имеются и другие, еще более редкие формы дистальной моторной нейропатии.
Причины
Из-за огромного разнообразия форм дистальной моторной нейропатии этиология нарушений при этом состоянии отличается большой вариативностью. Общей чертой является дегенерация моторных нейронов передних рогов спинного мозга или их отростков по тем или иным причинам. Например, наиболее изученная в этом плане дистальная моторная нейропатия 5-го типа обусловлена мутацией гена вSCL2, который локализован на 11-й хромосоме. Ген кодирует особый протеин сейпин, входящий в состав эндоплазматического ретикулума мотонейронов. Из-за мутации полученный белок имеет дефектную структуру, что затрудняет процессы его гликозилирования и утилизации, поэтому сейпин начинает накапливаться в нейронах. Дистальная моторная нейропатия 5-го типа развивается из-за того, что забитые комплексами сейпина нейроны подвергаются дистрофии и гибнут.
Схожие процессы имеют место и при других формах дистальной моторной нейропатии. По причине той или иной мутации происходит нарушение метаболизма или жизнедеятельности двигательных нейронов, из-за чего в последующем возникает заболевание. Поскольку для развития дистрофии нейронов или их волокон требуется неодинаковое время, при различных мутациях проявления патологии могут обнаруживаться вскоре после рождения, в детском, подростковом, взрослом или даже преклонном возрасте. Таким образом, совокупность всех форм дистальной моторной нейропатии создает непрерывный спектр заболеваний, способных поражать людей всех возрастов. Отличается и механизм наследования этих патологий - он может быть как аутосомно-доминантным, так и рецессивным. Существует как минимум одна форма такой нейропатии, сцепленная с Х-хромосомой.
Схожие процессы имеют место и при других формах дистальной моторной нейропатии. По причине той или иной мутации происходит нарушение метаболизма или жизнедеятельности двигательных нейронов, из-за чего в последующем возникает заболевание. Поскольку для развития дистрофии нейронов или их волокон требуется неодинаковое время, при различных мутациях проявления патологии могут обнаруживаться вскоре после рождения, в детском, подростковом, взрослом или даже преклонном возрасте. Таким образом, совокупность всех форм дистальной моторной нейропатии создает непрерывный спектр заболеваний, способных поражать людей всех возрастов. Отличается и механизм наследования этих патологий - он может быть как аутосомно-доминантным, так и рецессивным. Существует как минимум одна форма такой нейропатии, сцепленная с Х-хромосомой.
Классификация
По мнению многих врачей-генетиков, классификация форм этой патологии на сегодняшний момент пока далека от завершения. Для ряда типов дистальной моторной нейропатии еще не определены гены, ответственные за развитие патологического состояния. Во многих случаях остается неясным и его патогенез. Поэтому в настоящее время все известные формы этого заболевания принято делить по механизму наследования на аутосомно-доминантные, аутосомно-рецессивные и сцепленные с полом. Среди этих групп выделяют различные фенотипические подтипы дистальной моторной нейропатии, отличающиеся по клиническому течению. Названия подтипов происходят от английских терминов Hereditary motor neuropathy (HMN) или Distal spinal muscular atrophy (DSMA). Ниже представлены наиболее распространенные варианты дистальной моторной нейропатии.
1. HMN. Врожденный тип дистальной моторной нейропатии, проявляется уже у новорожденных детей. Характеризуется очень медленным прогрессированием, наследуется по аутосомно-доминантному механизму. Обусловлен дефектами гена TRPV4, локализованного на 12-й хромосоме.
1. HMN. Врожденный тип дистальной моторной нейропатии, проявляется уже у новорожденных детей. Характеризуется очень медленным прогрессированием, наследуется по аутосомно-доминантному механизму. Обусловлен дефектами гена TRPV4, локализованного на 12-й хромосоме.
Клиническая картина
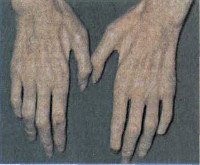
2. HMN2А. Дистальная моторная нейропатия типа 2А с аутосомно - доминантным характером наследования, обусловлена мутацией гена HSPB8, расположенного на 12 - й хромосоме - его же дефекты являются причиной другого типа нейропатии, известного как синдром Шарко - Мари - Тута. Возникает у взрослых людей, преимущественно поражаются верхние конечности. Характеризуется медленным нарастанием симптомов: мышечной слабости, атрофии и нарушений вибрационной чувствительности.
3. HMN2D. Дистальная моторная нейропатия с преимущественным поражением голеней. Вызвана мутацией гена FBXO38, который локализуется на 5-й хромосоме. Чаще всего первые симптомы возникают в подростковом или взрослом возрасте. В дебюте заболевания наблюдается слабость в коленях, которая затем перерастает в тремор нижних конечностей. На поздних этапах болезни в патологический процесс вовлекается мускулатура рук.
4. HMN5. Одна из самых часто встречающихся (относительно других типов заболевания) и поэтому наиболее изученная форма дистальной моторной нейропатии. Передается аутосомно-доминантно, причина патологии кроется в дефекте гена вSCL2, расположенного на 11-й хромосоме. Развивается преимущественно у подростков или взрослых людей, изредка у детей. В основном поражаются дистальные отделы рук, симптомы сводятся к развитию слабости и появлению тремора пальцев и кистей. Менее чем у половины больных в конечном итоге возникает слабость мышц дистальных отделов ног.
5. DSMA1. Дистальная спинальная амиотрофия 1 - го типа с аутосомно - рецессивным механизмом наследования. Обусловлена мутацией гена IGHMBP2, локализованного на 11-й хромосоме. Нередко характеризуется внутриутробным началом заболевания, наиболее сильно поражается мускулатура пояса верхних конечностей. Иногда дегенерация затрагивает двигательные нейроны, иннервирующие дыхательные мышцы, что резко усугубляет течение дистальной моторной нейропатии и может привести к смерти больного.
6. DSMAX. Форма дистальной моторной нейропатии, сцепленной с Х - хромосомой и обусловленной мутацией гена ATP7A. По причине сцепленного с полом наследования встречается у мальчиков, характеризуется ранним началом с поражением всех групп дистальных мышц конечностей.
Ассоциированные симптомы: Тремор.
3. HMN2D. Дистальная моторная нейропатия с преимущественным поражением голеней. Вызвана мутацией гена FBXO38, который локализуется на 5-й хромосоме. Чаще всего первые симптомы возникают в подростковом или взрослом возрасте. В дебюте заболевания наблюдается слабость в коленях, которая затем перерастает в тремор нижних конечностей. На поздних этапах болезни в патологический процесс вовлекается мускулатура рук.
4. HMN5. Одна из самых часто встречающихся (относительно других типов заболевания) и поэтому наиболее изученная форма дистальной моторной нейропатии. Передается аутосомно-доминантно, причина патологии кроется в дефекте гена вSCL2, расположенного на 11-й хромосоме. Развивается преимущественно у подростков или взрослых людей, изредка у детей. В основном поражаются дистальные отделы рук, симптомы сводятся к развитию слабости и появлению тремора пальцев и кистей. Менее чем у половины больных в конечном итоге возникает слабость мышц дистальных отделов ног.
5. DSMA1. Дистальная спинальная амиотрофия 1 - го типа с аутосомно - рецессивным механизмом наследования. Обусловлена мутацией гена IGHMBP2, локализованного на 11-й хромосоме. Нередко характеризуется внутриутробным началом заболевания, наиболее сильно поражается мускулатура пояса верхних конечностей. Иногда дегенерация затрагивает двигательные нейроны, иннервирующие дыхательные мышцы, что резко усугубляет течение дистальной моторной нейропатии и может привести к смерти больного.
6. DSMAX. Форма дистальной моторной нейропатии, сцепленной с Х - хромосомой и обусловленной мутацией гена ATP7A. По причине сцепленного с полом наследования встречается у мальчиков, характеризуется ранним началом с поражением всех групп дистальных мышц конечностей.
Ассоциированные симптомы: Тремор.
|
|
Диагностика
Для диагностики дистальной моторной нейропатии используют такие методы, как изучение наследственного анамнеза пациента, электронейромиографию и молекулярно-генетические исследования. При осмотре больного с практически любой формой этого состояния выявляется слабость мышц рук и/или ног, в ряде случаев регистрируется арефлексия. Если дистальная моторная нейропатия возникла достаточно давно, определяется гипотрофия мышц, а при тяжелом течении - атрофия мышечных тканей на дистальных участках конечностей. Изучение наследственного анамнеза в ряде случаев подтверждает аналогичные изменения у родителей или других близких родственников пациента.
Картина электромиографии (или электронейромиографии) может различаться в зависимости от типа дистальной моторной нейропатии и, следовательно, патогенеза этого состояния. Например, тип DSMA1 характеризуется достаточно заметным замедлением прохождения нервного импульса по волокнам, а для формы HMN5 типично нормальное значение этого показателя при отсутствии потенциала действия миоцитов. Биопсия мышечной ткани на пораженных участках практически всегда выявляет признаки гипотрофии и неоднородность миоцитов. Молекулярно-генетическая диагностика используется только для определения некоторых форм дистальной моторной нейропатии (например, HMN2А, HMN, DSMA1). При этом возможно проведение пренатальной диагностики таких состояний, материал для исследования получают методом биопсии ворсин хориона.
Картина электромиографии (или электронейромиографии) может различаться в зависимости от типа дистальной моторной нейропатии и, следовательно, патогенеза этого состояния. Например, тип DSMA1 характеризуется достаточно заметным замедлением прохождения нервного импульса по волокнам, а для формы HMN5 типично нормальное значение этого показателя при отсутствии потенциала действия миоцитов. Биопсия мышечной ткани на пораженных участках практически всегда выявляет признаки гипотрофии и неоднородность миоцитов. Молекулярно-генетическая диагностика используется только для определения некоторых форм дистальной моторной нейропатии (например, HMN2А, HMN, DSMA1). При этом возможно проведение пренатальной диагностики таких состояний, материал для исследования получают методом биопсии ворсин хориона.
Лечение
Специфического лечения ни одной из форм дистальной моторной нейропатии на сегодняшний день не существует. Поддерживающая терапия подразумевает использование ноотропных средств, препаратов, улучшающих трофику тканей, и витаминов, но эффективность таких мер крайне низка. Учитывая, что большинство форм дистальной моторной нейропатии характеризуются слабо выраженными симптомами и крайне медленным прогрессированием, некоторые специалисты предпочитают только наблюдать и контролировать развитие симптомов без назначения терапии. Физические упражнения, вопреки распространенному мнению, нередко не только не способствует замедлению развития заболевания, но и еще больше ускоряет дегенерацию нейронов или двигательных волокон. Профилактика дистальной моторной нейропатии возможна только в рамках пренатальной диагностики и медико-генетической консультации родителей перед зачатием ребенка.
Профилактика
Прогноз заболевания различается в зависимости от формы нейропатии. Врожденные и ювенильные разновидности, как правило, протекают тяжелее и могут стать причиной инвалидности в раннем возрасте. Кроме того, при этих формах в процесс иногда вовлекаются дыхательные мышцы, что существенно усугубляет течение дистальной моторной нейропатии. Разновидности заболевания, впервые проявляющиеся во взрослом возрасте, обычно намного легче протекают и медленнее прогрессируют. Однако и в этом случае всегда остается риск значительного ухудшения качества жизни больных из-за гипотрофии и слабости мышц верхних и нижних конечностей.