
ICD-10 codes
- ICD-10
- G71.0 Muscular dystrophy
Description
Окулофарингеальная мышечная дистрофия. Наследственное заболевание, которое имеет как аутосомно - доминантный, так и аутосомно - рецессивный вариант наследования, характеризуется преимущественным поражением мышц лица и головы. Симптомами этого состояния выступают расстройства глотания (дисфагия), птоз век, слабость мимических мышц и офтальмопарез. Иногда развивается общая мышечная слабость. Диагностика окулофарингеальной мышечной дистрофии производится на основании данных настоящего статуса пациента, электромиографии, молекулярно-генетических исследований. Специфического лечения патологии не существует, применяют паллиативные и симптоматические мероприятия.
Additional facts
Окулофарингеальная мышечная дистрофия (ОФМД) - генетическая патология, которая проявляется поражением мышц головы и отчасти конечностей и имеет особенный характер обуславливающей ее мутации. Исторически первой была открыта аутосомно-доминантная форма заболевания - в 1962-м году Виктор с командой коллег наблюдал у представителей трех семей необычное сочетание дисфагии с птозом век, симптомы со временем прогрессировали и приводили к слабости поясов верхних и нижних конечностей. Изучение наследственного анамнеза больных доказало аутосомно-доминантный механизм наследования окулофарингеальной мышечной дистрофии. Затем в 1975-м году японские исследователи Сатоёши и Киношита описали сходные симптомы миопатии у девочки, имеющей здоровых родителей - этот факт, а также более раннее развитие проявлений указывало на аутосомно-рецессивную разновидность ОФМД. На сегодняшний день точные цифры встречаемости заболевания не выяснены, отмечено только более частое возникновение в некоторых национальных группах (франкоязычные канадцы, бухарские евреи). По данным современной генетики, окулофарингеальная мышечная дистрофия с одинаковой частотой поражает как мужчин, так и женщин.
Reasons
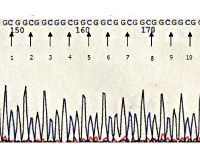
Уникальностью окулофарингеальной мышечной дистрофии является характер мутации, обуславливающей развитие этого наследственного заболевания. Дефект выявляется в гене PABPN1, который локализован на 14-й хромосоме. Он кодирует полиаденилат-связывающий белок, относящийся к группе протеинов клеточного ядра, который отвечает за стабилизацию образующихся в ядре молекул матричной РНК за счет их полиаденилирования. В структуре гена присутствует последовательность повторяющихся триплетов GCG, количество их повторов у здорового человека в 98% случаев равно шести. Именно изменение количества повторов данного триплета и обуславливает развитие всех форм окулофарингеальной мышечной дистрофии.
При наличии семи повторов GCG в гене PABPN1, особенно в гомозиготном состоянии, развивается аутосомно-рецессивная форма окулофарингеальной мышечной дистрофии. Однако такое происходит не всегда - примерно у 2% фенотипически здоровых людей наблюдается наличие семи повторов в этом гене, как в гетерозиготном, так и гомозиготном состоянии. Причины, почему у таких лиц не развивается ОФМД, достоверно не изучены. В случае более выраженного нарушения структуры гена PABPN1, при котором наблюдается свыше десяти повторов GCG, возникает аутосомно-доминантная разновидность окулофарингеальной мышечной дистрофии. При этом у гомозигот с таким нарушением (наличием такого количества повторов в обеих аллелях) патология протекает в целом тяжелее и диагностируется в среднем на 15-20 лет раньше.
Таким образом, особенностью окулофарингеальной мышечной дистрофии является тот факт, что это нарушение обусловлено «передозировкой» определенного генетического материала. При этом форма патологии в плане механизма ее наследования и симптомов зависит от масштабов вышеуказанной «передозировки». Наличие генетического дефекта приводит к появлению у полиаденилат-связывающего белка аномального «хвоста» из остатков аминокислоты аланина, что нарушает функции этого протеина. Именно с наличием полиаланиновой последовательности в структуре данного белка связывают появление нитевидных включений в ядрах клеток, которое является одним из признаков окулофарингеальной мышечной дистрофии.
При наличии семи повторов GCG в гене PABPN1, особенно в гомозиготном состоянии, развивается аутосомно-рецессивная форма окулофарингеальной мышечной дистрофии. Однако такое происходит не всегда - примерно у 2% фенотипически здоровых людей наблюдается наличие семи повторов в этом гене, как в гетерозиготном, так и гомозиготном состоянии. Причины, почему у таких лиц не развивается ОФМД, достоверно не изучены. В случае более выраженного нарушения структуры гена PABPN1, при котором наблюдается свыше десяти повторов GCG, возникает аутосомно-доминантная разновидность окулофарингеальной мышечной дистрофии. При этом у гомозигот с таким нарушением (наличием такого количества повторов в обеих аллелях) патология протекает в целом тяжелее и диагностируется в среднем на 15-20 лет раньше.
Таким образом, особенностью окулофарингеальной мышечной дистрофии является тот факт, что это нарушение обусловлено «передозировкой» определенного генетического материала. При этом форма патологии в плане механизма ее наследования и симптомов зависит от масштабов вышеуказанной «передозировки». Наличие генетического дефекта приводит к появлению у полиаденилат-связывающего белка аномального «хвоста» из остатков аминокислоты аланина, что нарушает функции этого протеина. Именно с наличием полиаланиновой последовательности в структуре данного белка связывают появление нитевидных включений в ядрах клеток, которое является одним из признаков окулофарингеальной мышечной дистрофии.
Symptoms
Клиническая картина окулофарингеальной мышечной дистрофии зависит от формы заболевания. Наиболее тяжелыми и ранними проявлениями характеризуется аутосомно-рецессивный тип этой патологии - нередко первые симптомы появляются еще в детском возрасте. Сначала регистрируется общая мышечная слабость, пониженная активность мимических мышц, незначительный птоз век. При аутосомно-рецессивной форме окулофарингеальной мышечной дистрофии достаточно быстро развиваются расстройства глотания - дисфагия, которая нередко делает процесс питания попросту невозможным. В дальнейшем возникает офтальмопарез и нарастающая мышечная дистрофия верхних и нижних конечностей.
Ассоциированные симптомы: Слабость мышц лица.
Ассоциированные симптомы: Слабость мышц лица.
Diagnostics
Для диагностики окулофарингеальной мышечной дистрофии применяются методики патогистологического изучения мышечных тканей, электромиографии и изучения наследственного анамнеза больного, а также молекулярно-генетические методы. При осмотре выявляют птоз век, расстройства глотания, общую мышечную слабость, снижение мимической активности. Возраст больных варьируется в зависимости от формы заболевания - при аутосомно-рецессивной разновидности пациентами в основном являются дети, в случае доминантного типа - взрослые 40-50-ти лет (реже 15-25-ти лет). Тщательный анализ наследственного анамнеза позволяет определить механизм наследования окулофарингеальной мышечной дистрофии.
При проведении электромиографии выявляют снижение амплитуды мышечных импульсов, их удлинение и полифазность. Биопсия мышечной ткани с последующим гистологическим изучением обычно обнаруживает наличие в ядрах клеток нитевидных включений, которые иногда приобретают разветвленный характер. При окулофарингеальной мышечной дистрофии нередко возникают нарушения в структуре волокон 1-го типа, при длительно текущем заболевании присоединяются признаки атрофии мышц. Молекулярно-генетическая диагностика этой патологии является надежнейшим и относительно простым (за счет характера мутации) методом определения. С этой целью врач-генетик производит амплификацию участка последовательности GCG с последующим электрофорезом для определения ее размера - увеличение количества повторов GCG указывает на наличие окулофарингеальной мышечной дистрофии. Также возможна пренатальная диагностика этого состояния генетическими методиками, материал для исследования получают методом амниоцентеза или биопсии ворсинок хориона.
При проведении электромиографии выявляют снижение амплитуды мышечных импульсов, их удлинение и полифазность. Биопсия мышечной ткани с последующим гистологическим изучением обычно обнаруживает наличие в ядрах клеток нитевидных включений, которые иногда приобретают разветвленный характер. При окулофарингеальной мышечной дистрофии нередко возникают нарушения в структуре волокон 1-го типа, при длительно текущем заболевании присоединяются признаки атрофии мышц. Молекулярно-генетическая диагностика этой патологии является надежнейшим и относительно простым (за счет характера мутации) методом определения. С этой целью врач-генетик производит амплификацию участка последовательности GCG с последующим электрофорезом для определения ее размера - увеличение количества повторов GCG указывает на наличие окулофарингеальной мышечной дистрофии. Также возможна пренатальная диагностика этого состояния генетическими методиками, материал для исследования получают методом амниоцентеза или биопсии ворсинок хориона.
Treatment
Специфического лечения окулофарингеальной мышечной дистрофии не существует, для уменьшения симптомов заболевания используют различные терапевтические и хирургические мероприятия. Для замедления прогрессирования патологии применяют преднизон, креатин и другие аналогичные препараты, однако их эффективность неодинакова у различных больных. Больным окулофарингеальной мышечной дистрофией назначают разнообразные физические упражнения, которые снижают скорость нарастания мышечной слабости. При выраженном птозе век и связанном с ним нарушении зрения можно использовать специальный скотч для поддержки или же, при относительно сохраненной активности мимических мышц, производить хирургическую коррекцию (блефаропластику).
Наиболее тяжелым осложнением окулофарингеальной мышечной дистрофии, требующим медицинского вмешательства, является дисфагия, в тяжелых случаях приводящая к полной невозможности глотания. В такой ситуации применяют назогастральные или другие зонды для питания больного, иногда накладывают стому. Хирургические методы облегчения симптомов дисфагии (например, рассечение перстнеглоточной мышцы) могут привести к временному облегчению, но в настоящий момент большинство специалистов стараются не использовать такие методики.
Наиболее тяжелым осложнением окулофарингеальной мышечной дистрофии, требующим медицинского вмешательства, является дисфагия, в тяжелых случаях приводящая к полной невозможности глотания. В такой ситуации применяют назогастральные или другие зонды для питания больного, иногда накладывают стому. Хирургические методы облегчения симптомов дисфагии (например, рассечение перстнеглоточной мышцы) могут привести к временному облегчению, но в настоящий момент большинство специалистов стараются не использовать такие методики.