
ICD-10 codes
- ICD-10
- H53.0 Amblyopia ex anopsia
Description
Амавроз Лебера. Наследственное заболевание, характеризующееся врожденным поражением светочувствительных клеток сетчатки глаза и в некоторых случаях другими общими нарушениями (аномалии почек, ЦНС). При этой патологии в первые месяцы жизни ребенка или сразу после рождения появляется нистагм, ослабление или отсутствие реакции зрачка на свет. В дальнейшем ребенок может тереть глаза (симптом Франческетти), возникает дальнозоркость и светобоязнь, возможна полная потеря зрения. Диагностика основывается на данных осмотра пациента врачом-офтальмологом, электроретинографии, исследования наследственного анамнеза и генетических анализов. Специфическое лечение амавроза Лебера на сегодняшний день не разработано.
Additional facts
Врожденный амавроз Лебера представляет собой гетерогенную группу заболеваний, причиной которых выступают мутации в 18 генах, кодирующих различные белки сетчатки, в том числе опсин. Впервые амавроз был описан еще в XIX веке (в 1867 году) Т. Лебером, указавшим основные проявления этого заболевания - маятниковый нистагм, слепота, появление пигментных пятен и включений на глазном дне. Средняя распространенность заболевания составляет 3:100000 населения. Основной механизм наследования заболевания - аутосомно-рецессивный, но есть также формы, передающиеся по аутосомно-доминантному принципу. Амавроз Лебера в равной мере поражает как мужчин, так и женщин. Заболевание составляет примерно 5% от всех наследственных ретинопатий. Современная генетика разрабатывает методики лечения данной патологии, имеются обнадеживающие результаты генной терапии одной из форм амавроза Лебера, обусловленной мутацией в гене RPE65.
Отдельно выделяют атрофию зрительных нервов Лебера, которая также характеризуется постепенной потерей остроты зрения и впоследствии полной слепотой. Однако это заболевание совершенно другой генетической природы и обусловлено повреждением митохондриальной ДНК, которая имеет свой уникальный тип наследования (по материнской линии).
Отдельно выделяют атрофию зрительных нервов Лебера, которая также характеризуется постепенной потерей остроты зрения и впоследствии полной слепотой. Однако это заболевание совершенно другой генетической природы и обусловлено повреждением митохондриальной ДНК, которая имеет свой уникальный тип наследования (по материнской линии).
Reasons
Основной механизм расстройства зрения при амаврозе Лебера - нарушение метаболизма в палочках и колбочках, которое ведет к летальным повреждениям фоторецепторов и их разрушению. Однако непосредственная причина таких изменений различается в зависимости от того, мутация какого именно гена вызвала заболевание.
Один из наиболее распространенных типов амавроза Лебера (тип 2, LCA2) обусловлен наличием мутантного гена RPE65 на первой хромосоме. Известно более 80-ти мутаций этого гена, некоторые из которых, помимо амавроза Лебера, вызывают и определенные формы пигментной абиотрофии сетчатки. Белок, кодируемый PRE65, отвечает за метаболизм ретинола в пигментном эпителии сетчатой оболочки глаза, поэтому при наличии генетического дефекта этот процесс нарушается с развитием побочных метаболических путей. В результате этого синтез родопсина в фоторецепторах прекращается, что и приводит к характерной клинической картине заболевания. Мутантные формы гена наследуются по аутосомно-рецессивному механизму.
Менее распространенная форма амавроза Лебера (тип 14) вызвана мутацией гена LRAT на 4-й хромосоме. Он кодирует белок лецитин-ретинол-ацилтрансферазу, который располагается в микросомах гепатоцитов и обнаружен в сетчатке глаза. Этот фермент участвует в метаболизме ретиноидов и витамина А, из-за наличия мутаций в гене полученный протеин не может полноценно выполнять свои функции, из-за чего развивается дегенерация фоторецепторов, которая клинически проявляется амаврозом Лебера или ювенильной пигментной абиотрофией сетчатки. Имеет аутосомно-рецессивный характер наследования.
Амавроз Лебера тип 8 наиболее часто приводит к врожденной слепоте, ответственный за развитие этой формы заболевания ген сRB1 располагается на 1-й хромосоме и имеет аутосомно-рецессивный характер наследования. При этом выяснено, что кодируемый данным геном белок принимает непосредственное участие в эмбриональном развитии фоторецепторов и пигментного эпителия сетчатки. Более точных данных по патогенезу данной формы амавроза Лебера на сегодняшний день не накоплено. Аналогичная ситуация с мутацией гена LCA5, расположенного в 6-й хромосоме и ассоциированного с 5-м типом амавроза. В настоящее время выявлен только белок, кодируемым данным геном - леберцилин, но его функции в сетчатке непонятны.
Также выявлено две формы амавроза Лебера, которые наследуются по аутосомно-доминантному механизму - тип 7, обусловленный мутацией гена сRX, и тип 11, ассоциированный с нарушением гена IMPDH1. Ген сRX кодирует белок, который обладает множеством функций - контроль развития фоторецепторов в эмбриональный период, поддержание их адекватного уровня во взрослом возрасте, участие в синтезе других протеинов сетчатки (является фактором транскрипции). Поэтому в зависимости от характера мутации гена сRX клиника амавроза Лебера 7-го типа может быть разнообразной - от врожденной слепоты до относительно позднего и вялотекущего ухудшения зрения. Инозин-5 -монофосфатдегидрогеназа 1, кодируемый геном IMPDH1, представляет собой фермент, регулирующий рост клеток и образование нуклеиновых кислот, однако это пока не позволяет прояснить патогенез того, как нарушения этого белка приводят к 11-му типу амавроза Лебера.
Один из наиболее распространенных типов амавроза Лебера (тип 2, LCA2) обусловлен наличием мутантного гена RPE65 на первой хромосоме. Известно более 80-ти мутаций этого гена, некоторые из которых, помимо амавроза Лебера, вызывают и определенные формы пигментной абиотрофии сетчатки. Белок, кодируемый PRE65, отвечает за метаболизм ретинола в пигментном эпителии сетчатой оболочки глаза, поэтому при наличии генетического дефекта этот процесс нарушается с развитием побочных метаболических путей. В результате этого синтез родопсина в фоторецепторах прекращается, что и приводит к характерной клинической картине заболевания. Мутантные формы гена наследуются по аутосомно-рецессивному механизму.
Менее распространенная форма амавроза Лебера (тип 14) вызвана мутацией гена LRAT на 4-й хромосоме. Он кодирует белок лецитин-ретинол-ацилтрансферазу, который располагается в микросомах гепатоцитов и обнаружен в сетчатке глаза. Этот фермент участвует в метаболизме ретиноидов и витамина А, из-за наличия мутаций в гене полученный протеин не может полноценно выполнять свои функции, из-за чего развивается дегенерация фоторецепторов, которая клинически проявляется амаврозом Лебера или ювенильной пигментной абиотрофией сетчатки. Имеет аутосомно-рецессивный характер наследования.
Амавроз Лебера тип 8 наиболее часто приводит к врожденной слепоте, ответственный за развитие этой формы заболевания ген сRB1 располагается на 1-й хромосоме и имеет аутосомно-рецессивный характер наследования. При этом выяснено, что кодируемый данным геном белок принимает непосредственное участие в эмбриональном развитии фоторецепторов и пигментного эпителия сетчатки. Более точных данных по патогенезу данной формы амавроза Лебера на сегодняшний день не накоплено. Аналогичная ситуация с мутацией гена LCA5, расположенного в 6-й хромосоме и ассоциированного с 5-м типом амавроза. В настоящее время выявлен только белок, кодируемым данным геном - леберцилин, но его функции в сетчатке непонятны.
Также выявлено две формы амавроза Лебера, которые наследуются по аутосомно-доминантному механизму - тип 7, обусловленный мутацией гена сRX, и тип 11, ассоциированный с нарушением гена IMPDH1. Ген сRX кодирует белок, который обладает множеством функций - контроль развития фоторецепторов в эмбриональный период, поддержание их адекватного уровня во взрослом возрасте, участие в синтезе других протеинов сетчатки (является фактором транскрипции). Поэтому в зависимости от характера мутации гена сRX клиника амавроза Лебера 7-го типа может быть разнообразной - от врожденной слепоты до относительно позднего и вялотекущего ухудшения зрения. Инозин-5 -монофосфатдегидрогеназа 1, кодируемый геном IMPDH1, представляет собой фермент, регулирующий рост клеток и образование нуклеиновых кислот, однако это пока не позволяет прояснить патогенез того, как нарушения этого белка приводят к 11-му типу амавроза Лебера.
Classification
В настоящее время полностью доказана взаимосвязь между клиническими проявлениями и мутациями определенных генов для 16-ти типов амавроза Лебера. Также имеются указания об открытии еще двух генов, повреждения в которых приводят к такому заболеванию, но пока в этом отношении проводятся дополнительные исследования.
• Тип 1 (LCA1, от английского Leber’s congenital amaurosis) - поврежденный ген GUCY2D на 17-й хромосоме, тип наследования аутосомно-рецессивный.
• Тип 2 (LCA2). Поврежденный ген RPE65 на 1 - й хромосоме, аутосомно - рецессивное наследование, имеются первые положительные результаты по генной терапии этой формы амавроза Лебера.
• Тип 3 (LCA3). Поврежденный ген RDH12 на 14 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 4 (LCA4). Поврежденный ген AIPL1 на 17 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 5 (LCA5). Поврежденный ген LCA5 на 6 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 6 (LCA6). Поврежденный ген RPGRIP1 на 14 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 7 (LCA7). Поврежденный ген сRX на 19 - й хромосоме, аутосомно - доминантное наследование. Характеризуется вариабельной клинической картиной.
• Тип 8 (LCA8). Поврежденный ген сRB1 на 1 - й хромосоме, аутосомно - рецессивное наследование. Статистически чаще остальных типов приводит к врожденной слепоте.
• Тип 9 (LCA9). Поврежденный ген LCA9 на 1 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 10 (LCA10). Поврежденный ген сEP290 на 12 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 11 (LCA11). Поврежденный ген IMPDH1 на 7 - й хромосоме, аутосомно - доминантное наследование.
• Тип 12 (LCA12). Поврежденный ген RD3 на 1 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 13 (LCA13). Поврежденный ген RDH12 на 14 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 14 (LCA14). Поврежденный ген LRAT на 4 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 15 (LCA15). Поврежденный ген TULP1 на 6 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 16 (LCA16). Поврежденный ген KCNJ13 на 2 - й хромосоме, аутосомно - рецессивное наследование.
Кроме того, иногда в клинической классификации выделяют не только название поврежденного гена, но и характер мутации, поскольку это имеет значительное влияние на течение амавроза Лебера. Более того, различные типы мутаций в одном и том же гене могут приводить к совершенно разным заболеваниям - например, некоторые разновидности делеций в гене сRX могут приводить не к амаврозу, а к палочко-колбочковой дистрофии. Некоторые мутации генов RPE65, LRAT и сRB1 являются причиной различных форм пигментной абиотрофии сетчатки.
• Тип 1 (LCA1, от английского Leber’s congenital amaurosis) - поврежденный ген GUCY2D на 17-й хромосоме, тип наследования аутосомно-рецессивный.
• Тип 2 (LCA2). Поврежденный ген RPE65 на 1 - й хромосоме, аутосомно - рецессивное наследование, имеются первые положительные результаты по генной терапии этой формы амавроза Лебера.
• Тип 3 (LCA3). Поврежденный ген RDH12 на 14 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 4 (LCA4). Поврежденный ген AIPL1 на 17 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 5 (LCA5). Поврежденный ген LCA5 на 6 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 6 (LCA6). Поврежденный ген RPGRIP1 на 14 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 7 (LCA7). Поврежденный ген сRX на 19 - й хромосоме, аутосомно - доминантное наследование. Характеризуется вариабельной клинической картиной.
• Тип 8 (LCA8). Поврежденный ген сRB1 на 1 - й хромосоме, аутосомно - рецессивное наследование. Статистически чаще остальных типов приводит к врожденной слепоте.
• Тип 9 (LCA9). Поврежденный ген LCA9 на 1 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 10 (LCA10). Поврежденный ген сEP290 на 12 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 11 (LCA11). Поврежденный ген IMPDH1 на 7 - й хромосоме, аутосомно - доминантное наследование.
• Тип 12 (LCA12). Поврежденный ген RD3 на 1 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 13 (LCA13). Поврежденный ген RDH12 на 14 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 14 (LCA14). Поврежденный ген LRAT на 4 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 15 (LCA15). Поврежденный ген TULP1 на 6 - й хромосоме, аутосомно - рецессивное наследование.
• Тип 16 (LCA16). Поврежденный ген KCNJ13 на 2 - й хромосоме, аутосомно - рецессивное наследование.
Кроме того, иногда в клинической классификации выделяют не только название поврежденного гена, но и характер мутации, поскольку это имеет значительное влияние на течение амавроза Лебера. Более того, различные типы мутаций в одном и том же гене могут приводить к совершенно разным заболеваниям - например, некоторые разновидности делеций в гене сRX могут приводить не к амаврозу, а к палочко-колбочковой дистрофии. Некоторые мутации генов RPE65, LRAT и сRB1 являются причиной различных форм пигментной абиотрофии сетчатки.
Symptoms
Симптоматика амавроза Лебера достаточно вариабельна и зависит от типа заболевания и характера мутации гена. В большинстве случаев при рождении ребенка патология не определяется - даже при осмотре глазного дна изменения наблюдаются лишь в нескольких процентах случаев. По мере его роста родители могут замечать, что ребенок не задерживает взгляд на предметах и окружающих, а в более старшем возрасте может болезненно реагировать на свет (появляется фотофобия), часто тереть глаза и указывать на них пальцем (симптом Франческетти, окулопальцевый синдром). Обнаруживается нистагм, который возникает еще в первые 2-3 месяца жизни и часто является одним из первых проявлений амавроза Лебера, замедленная реакция зрачка на свет или ее полное отсутствие.
Diagnostics
В современной офтальмологии диагностика амавроза Лебера производится на основании осмотра глазного дна, мониторинга динамики изменений в нем, данных электроретинографии. Немаловажную роль играет также изучение наследственного анамнеза, а для некоторых типов заболевания - генетическое секвенирование последовательности ключевых генов.
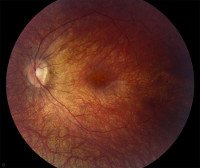
При осмотре глазного дна относительно долгое время (первые несколько лет жизни) никаких изменений может не регистрироваться. Первыми, но не специфическими офтальмологическими симптомами амавроза являются нистагм, косоглазие, замедленная или отсутствующая реакция зрачков на свет. Возникающие со временем изменения сетчатки сводятся к появлению пигментных или непигментированных пятен различного размера, сужения артериол, бледности диска зрительного нерва. К 8-10 годам практически у всех больных наблюдаются костные пигментные тельца, расположенные по периферии глазного дна. Характерным признаком является более быстрое прогрессирование изменений на сетчатке по сравнению с функциональными нарушениями зрения, которые развиваются относительно медленно. До развития слепоты острота зрения составляет 0,1 и менее, часто регистрируется дальнозоркость, светобоязнь.
У подростков и взрослых людей помимо указанных симптомов могут диагностироваться кератоконус и катаракта. Электроретинография при амаврозе Лебера, как правило, отражает сильное снижение амплитуды всех волн или их полное отсутствие. Генетические исследования позволяют выявить поврежденный ген и тип мутации только в 50-60% случаев (частота наиболее распространенных повреждений генов). Подавляющее большинство клиник производят секвенирование последовательностей с целью выявления мутаций только в отношении генов RPE65, сRX, сRB1, LCA5 и KCNJ13.
При осмотре глазного дна относительно долгое время (первые несколько лет жизни) никаких изменений может не регистрироваться. Первыми, но не специфическими офтальмологическими симптомами амавроза являются нистагм, косоглазие, замедленная или отсутствующая реакция зрачков на свет. Возникающие со временем изменения сетчатки сводятся к появлению пигментных или непигментированных пятен различного размера, сужения артериол, бледности диска зрительного нерва. К 8-10 годам практически у всех больных наблюдаются костные пигментные тельца, расположенные по периферии глазного дна. Характерным признаком является более быстрое прогрессирование изменений на сетчатке по сравнению с функциональными нарушениями зрения, которые развиваются относительно медленно. До развития слепоты острота зрения составляет 0,1 и менее, часто регистрируется дальнозоркость, светобоязнь.
У подростков и взрослых людей помимо указанных симптомов могут диагностироваться кератоконус и катаракта. Электроретинография при амаврозе Лебера, как правило, отражает сильное снижение амплитуды всех волн или их полное отсутствие. Генетические исследования позволяют выявить поврежденный ген и тип мутации только в 50-60% случаев (частота наиболее распространенных повреждений генов). Подавляющее большинство клиник производят секвенирование последовательностей с целью выявления мутаций только в отношении генов RPE65, сRX, сRB1, LCA5 и KCNJ13.
Differential diagnosis
Дифференциальную диагностику производят с различными формами пигментной абиотрофии сетчатки (при ней сохраняется нормальная или немного сниженная амплитуда волн на электроретинограмме) и некоторыми типами атрофии зрительных нервов.
Treatment
На сегодняшний день специфического лечения любого типа амавроза Лебера не существует. На этапе клинических испытаний находится генно-инженерное введение гена RPE65 в сетчатую оболочку глаза больных амаврозом 2-го типа, имеются первые данные о значительном улучшении зрения подопытных больных. В случае же остальных форм заболевания такого прогресса пока нет. Поддерживающее лечение сводится к витаминной терапии, внутриглазным инъекциям сосудорасширяющих средств. При дальнозоркости назначается ношение очков.
Forecast
В плане сохранения зрения прогноз крайне неблагоприятный, практически 95% больных полностью теряют способность видеть к 10-му году жизни. Кроме того, это наследственное заболевание может осложняться проблемами с ЦНС, почками, эндокринной системой, что требует более тщательного медицинского мониторинга для своевременного выявления подобных нарушений.
Similar diseases
Related standards of medical care
Basic medical services (according to which clinics are selected)
Medical consultations
- Consultation with a physiotherapist
- Consultation of an ophthalmologist
Non-surgical treatment
- Massage of the cervical-collar zone
- Local magnetotherapy
Clinics with the best prices (of 14 selected medical services)
|
| МЦ Доктор на проспекте Энтузиастов - Санкт-Петербург (M. Ладожская) | +7(499..show+7(499) 116-82-39 |
rating: 4.5
|
9800₽ | |
| Мама, Папа, Я в Люберцах - Люберцы (M. Жулебино) | +7(495..show+7(499) 116-82-39 +7(495) 150-33-88 |
rating: 4.4
|
10620₽ | |
| Мама, Папа, Я на Академика Королева - Москва (M. ВДНХ) | +7(499..show+7(499) 116-82-39 +7(499) 116-77-93 +7(495) 150-33-88 |
rating: 4.4
|
10650₽ | |
| Мама, Папа, Я на Барклая - Москва (M. Багратионовская) | +7(495..show+7(499) 116-82-39 +7(495) 150-33-88 |
rating: 4.4
|
10650₽ | |
| МЕДСИ в Митино - Москва (M. Пятницкое шоссе) | +7(495..show+7(499) 116-82-39 +7(495) 023-60-84 |
rating: 4.2
|
11110₽ | |
| ЦКБ Гражданской авиации на Иваньковском шоссе - Москва (M. Сокол) | +7(495..show+7(499) 116-82-39 +7(495) 490-03-78 +7(495) 490-01-46 +7(495) 490-04-90 +7(495) 490-04-37 |
rating: 4.6
|
11362₽ | |
| МЕДСИ в Хорошевском проезде - Москва (M. Беговая) | +7(495..show+7(499) 116-82-39 +7(495) 780-40-64 +7(495) 152-55-46 |
rating: 4.3
|
11880₽ | |
| МЕДСИ на Рублевском шоссе - Москва (M. Кунцевская) | +7(495..show+7(499) 116-82-39 +7(495) 023-60-84 |
rating: 4.4
|
12110₽ | |
| МЕДСИ на Ленинградском проспекте - Москва (M. Аэропорт) | +7(495..show+7(499) 116-82-39 +7(495) 229-18-75 +7(495) 152-55-46 |
rating: 4.4
|
12110₽ | |
| МЕДСИ в Красногорске - Красногорск (M. Пятницкое шоссе) | +7(495..show+7(499) 116-82-39 +7(495) 730-57-15 +7(495) 152-55-46 |
rating: 4.4
|
12110₽ | |