Другие названия и синонимы
Ahlstrom syndrome.
МКБ-10 коды
|
|
Описание
Синдром Альстрома. Это генетическая патология, при которой развиваются ожирение, пигментный ретинит, сахарный диабет 2 типа, нейросенсорная тугоухость, болезни сердца и нефропатия. Уже в первом десятилетии жизни отмечается постепенное снижение зрения и слуха, присоединяются новые инсулинорезистентность, повышенное артериальное давление, сердечная недостаточность, протеинурия, гипогонадизм. Для подтверждения диагноза проводится ЭхоС, офтальмоскопия, ЭРГ, аудиометрия, биохимия крови, ДНК-диагностика. Симптоматическое лечение: диета, сахароснижающие препараты, ингибиторы АПФ, выбор слухового аппарата, тонированные очки.
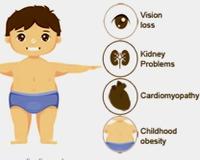
Дополнительные факты
Синдром Альстрома - редкое генетическое заболевание с низкой популяционной частотой (менее 1 случая на 1000000 населения). В настоящее время в международном регистре зарегистрировано более 1050 таких пациентов из более чем 60 стран мира. Описаны случаи рождения 2-3 детей с этим заболеванием в одной семье. Патология стала известна и получила свое название в 1959 году благодаря шведскому офтальмологу Альстрёму, молекулярно-генетическая основа синдрома была открыта в 1997 году. Клиническое значение генетического нарушения заключается в раннем начале и развитии тяжелых системных осложнений.
Причины
Синдром Альстрома связан с аутосомно-рецессивным наследованием дефектных копий генов от обоих родителей, бессимптомных носителей мутаций. Ген заболевания - ALMS1 - расположен на коротком плече 2-й хромосомы в локусе 2p13. Продуктом этого гена является одноименный белок, который участвует в образовании микротрубочек и ресничек, транспорте веществ внутри клетки. Синдром Альстрома относится к группе цилиопатий. В настоящее время описано несколько десятков мутаций гена ALMS1, которые в гомозиготном состоянии вызывают развитие генетического заболевания.
Патогенез
Ген ALMS1 экспрессируется во многих тканях организма: в большей степени в гонадах, головном мозге и сетчатке глаза и в меньшей степени в почках, селезенке и печени. Его мутации приводят к синтезу дефектного рецепторного белка, который вызывает структурные и функциональные аномалии ресничек - ресничек. В результате реснички лишаются способности выполнять сенсорные и моторные функции, особенно в тех органах, в которых наблюдается повышенная экспрессия ALMS1.
Предполагается, что выработка аномального белка в палочках и колбочках сетчатки вызывает инфильтрацию пигмента, дегенерацию и атрофию нейронепителия. Дисфункция гонад сопровождается развитием гипогонадотропного гипогонадизма, поджелудочная железа - нарушением толерантности к глюкозе. Повреждение клеточных структур головного мозга, в частности обонятельной луковицы, приводит к перееданию. Механизм развития некоторых нарушений при синдроме Альстрома остается неясным.
Предполагается, что выработка аномального белка в палочках и колбочках сетчатки вызывает инфильтрацию пигмента, дегенерацию и атрофию нейронепителия. Дисфункция гонад сопровождается развитием гипогонадотропного гипогонадизма, поджелудочная железа - нарушением толерантности к глюкозе. Повреждение клеточных структур головного мозга, в частности обонятельной луковицы, приводит к перееданию. Механизм развития некоторых нарушений при синдроме Альстрома остается неясным.
Клиническая картина
Большинство клинических признаков синдрома Альсстрема проявляются в раннем детстве, другая часть - в течение первого или второго десятилетия жизни. Самые ранние симптомы - светобоязнь и нистагм - появляются через 6-15 месяцев и сигнализируют о прогрессирующей дистрофии сетчатки. Дети с синдромом Альсстрема рождаются с нормальной массой тела, но ожирение развивается ближе к 1-2 годам. Потеря слуха регистрируется примерно с 5 лет. Дилатационная кардиомиопатия может возникнуть в любом возрасте и на любой стадии заболевания.
В возрасте от 1,5 до 5 лет развивается инсулинорезистентность, которая превращается в сахарный диабет 2 типа в возрасте от 20 до 30 лет. Характерны и другие эндокринные нарушения: гипотиреоз, у мальчиков - гипогонадотропный гипогонадизм, крипторхизм, у девочек - аменорея, гирсутизм, СПКЯ, позже - эндометриоз. Во второй декаде развиваются гепатолический синдром, нефропатия и макропротеинурия. Возможно недержание мочи.
Рост ребенка до полового созревания на 1-3 года опережает костный возраст, однако из-за раннего закрытия зон роста окончательный рост обычно ниже, чем в популяции. Из аномалий скелета часто присутствуют нарушения осанки, брахидактилия. Выявлены дефекты верхнего зубного ряда. Из кожных проявлений наиболее типичны черный акантоз и алопеция. Интеллект при синдроме Альстрома, как правило, не снижен, у некоторых пациентов описана ХБП.
Ассоциированные симптомы: Гиперурикемия. Изменение веса. Повышенное АД. Светобоязнь. Увеличение веса.
В возрасте от 1,5 до 5 лет развивается инсулинорезистентность, которая превращается в сахарный диабет 2 типа в возрасте от 20 до 30 лет. Характерны и другие эндокринные нарушения: гипотиреоз, у мальчиков - гипогонадотропный гипогонадизм, крипторхизм, у девочек - аменорея, гирсутизм, СПКЯ, позже - эндометриоз. Во второй декаде развиваются гепатолический синдром, нефропатия и макропротеинурия. Возможно недержание мочи.
Рост ребенка до полового созревания на 1-3 года опережает костный возраст, однако из-за раннего закрытия зон роста окончательный рост обычно ниже, чем в популяции. Из аномалий скелета часто присутствуют нарушения осанки, брахидактилия. Выявлены дефекты верхнего зубного ряда. Из кожных проявлений наиболее типичны черный акантоз и алопеция. Интеллект при синдроме Альстрома, как правило, не снижен, у некоторых пациентов описана ХБП.
Ассоциированные симптомы: Гиперурикемия. Изменение веса. Повышенное АД. Светобоязнь. Увеличение веса.
Возможные осложнения
Прогрессирование пигментного ретинита приводит к полной слепоте к 7-8 годам. Возможно развитие гипергликемии, диабетического кетоацидоза. Все пациенты бесплодны. В любом возрасте высок риск внезапной смерти от сердечной недостаточности из-за кардиомиопатии. Почечная недостаточность - причина смерти в зрелом возрасте. Продолжительность жизни обычно не превышает 40 лет.
Диагностика
К ведению пациентов с синдромом Альстрома привлекаются врачи разных специальностей: педиатры, офтальмологи, аудиологи, эндокринологи, кардиологи. При физикальном обследовании у пациентов отмечается повышенный ИМТ, чрезмерное развитие подкожно-жирового слоя, гиперпигментация кожи в складках тела, часто - артериальная гипертензия. Для уточнения диагностической гипотезы выполняется следующее:
• Офтальмологическая диагностика. По данным визометрии диагностируется снижение остроты зрения. При осмотре глазного дна обнаруживаются признаки дегенерации пигмента сетчатки. Электроретинограмма демонстрирует ослабление амплитуды биоэлектрических волн.
• Сурдологическое обследование. Аудиометрическое обследование слуха выявляет двустороннюю потерю слуха от умеренной до тяжелой. При планировании слуховых аппаратов проводится измерение акустического импеданса, ОАЭ, электрокохлеография.
• Осмотр внутренних органов. По результатам УЗИ ОБП определяется гепатоспленомегалия, жировой гепатоз. Для определения размеров и функции сердца, своевременного выявления ДКМП назначают ЭхоКГ. У подростков с синдромом Альстрома рекомендуется ультразвуковое исследование половых органов (органов малого таза, мошонки) для оценки состояния гонад.
• лабораторные образцы. В биохимическом анализе крови определяется повышенный уровень трансаминаз, триглицеридов, инсулина, глюкозы и гликозилированного гемоглобина. В моче регистрируются глюкозурия, протеинурия, гиперурикемия. Для оценки других эндокринных нарушений определяется уровень тиреоидных, гонадотропных и половых гормонов.
• Генодиагностика. Обследованы и родители, и больной ребенок. Молекулярно-генетический анализ гена ALMS1 показывает гетерозиготную передачу мутации у матери от отца и гомозиготную у ребенка.
• Офтальмологическая диагностика. По данным визометрии диагностируется снижение остроты зрения. При осмотре глазного дна обнаруживаются признаки дегенерации пигмента сетчатки. Электроретинограмма демонстрирует ослабление амплитуды биоэлектрических волн.
• Сурдологическое обследование. Аудиометрическое обследование слуха выявляет двустороннюю потерю слуха от умеренной до тяжелой. При планировании слуховых аппаратов проводится измерение акустического импеданса, ОАЭ, электрокохлеография.
• Осмотр внутренних органов. По результатам УЗИ ОБП определяется гепатоспленомегалия, жировой гепатоз. Для определения размеров и функции сердца, своевременного выявления ДКМП назначают ЭхоКГ. У подростков с синдромом Альстрома рекомендуется ультразвуковое исследование половых органов (органов малого таза, мошонки) для оценки состояния гонад.
• лабораторные образцы. В биохимическом анализе крови определяется повышенный уровень трансаминаз, триглицеридов, инсулина, глюкозы и гликозилированного гемоглобина. В моче регистрируются глюкозурия, протеинурия, гиперурикемия. Для оценки других эндокринных нарушений определяется уровень тиреоидных, гонадотропных и половых гормонов.
• Генодиагностика. Обследованы и родители, и больной ребенок. Молекулярно-генетический анализ гена ALMS1 показывает гетерозиготную передачу мутации у матери от отца и гомозиготную у ребенка.
Диф. диагностика
Синдром Альстрома необходимо отличать от других генетических и врожденных патологий, которые возникают при нарушении обмена веществ, пигментном ретините и дисфункции внутренних органов:
• синдром Барде-Бидла;
• амаврозы Лебера;
• первичный DCMP;
• Синдром Прадера-Вилли.
• синдром Барде-Бидла;
• амаврозы Лебера;
• первичный DCMP;
• Синдром Прадера-Вилли.
|
|
Лечение
Коррекция патологии требует привлечения междисциплинарной команды специалистов. Из-за отсутствия специфического лечения возможно проведение только симптоматической терапии. Используются поведенческие, лечебные, хирургические методы:
• при ожирении. Диетотерапия, регулярные физические нагрузки;
• при сахарном диабете. Прием гипогликемических средств, инсулинотерапия;
• при дилатационной кардиомиопатии - ингибиторы АПФ, диуретики, сердечные гликозиды, в терминальной стадии сердечной недостаточности - трансплантация сердца;
• при гиперлипидемии. Статины;
• с нейросенсорной тугоухостью. Слуховые аппараты с цифровыми слуховыми аппаратами, кохлеарные имплантаты;
• с светобоязнью. Ношение затемненных очков.
• при ожирении. Диетотерапия, регулярные физические нагрузки;
• при сахарном диабете. Прием гипогликемических средств, инсулинотерапия;
• при дилатационной кардиомиопатии - ингибиторы АПФ, диуретики, сердечные гликозиды, в терминальной стадии сердечной недостаточности - трансплантация сердца;
• при гиперлипидемии. Статины;
• с нейросенсорной тугоухостью. Слуховые аппараты с цифровыми слуховыми аппаратами, кохлеарные имплантаты;
• с светобоязнью. Ношение затемненных очков.
Прогноз
Продолжительность жизни пациентов с синдромом Альстрома сокращается. Причины смерти - тяжелые метаболические осложнения, сердечная и почечная недостаточность. Систематическое наблюдение и выполнение всех врачебных предписаний позволяет добиться компенсации симптомов, улучшить качество и продлить жизнь. Профилактика заключается в исключении близкородственных браков и предоставлении генетических консультаций семьям с синдромом Альстрома.
Список литературы
1. Синдром Альстрема у подростка (клиническое наблюдение)/ Оленев А. С., Тыртова Л. В., Паршина Н. В. Педиатр. 2017.
2. Синдром Альстрема у подростков (первое описание в России)/ Щербачева Л.Н., Цитлидзе Н.М., Смирнова Г.Е., Кураева Т.Л., Бровкин А.Н. Сахарный диабет. 2007.
3. Молекулярная генетика и клинические особенности моногенных форм сахарного диабета/ Петеркова В. А., Кураева Т. Л. и // Вестник Российской академии медицинских наук. 2012.
2. Синдром Альстрема у подростков (первое описание в России)/ Щербачева Л.Н., Цитлидзе Н.М., Смирнова Г.Е., Кураева Т.Л., Бровкин А.Н. Сахарный диабет. 2007.
3. Молекулярная генетика и клинические особенности моногенных форм сахарного диабета/ Петеркова В. А., Кураева Т. Л. и // Вестник Российской академии медицинских наук. 2012.