Другие названия и синонимы
Usher syndrome, Синдром Ашера.
Описание
Синдром Ашера. Это редкое генетическое заболевание, связанное с врожденной нейросенсорной тугоухостью, прогрессирующим пигментным ретинитом и вестибулярной атаксией. В зависимости от типа синдрома у пациентов наблюдаются следующие симптомы: значительная потеря слуха или глухота, снижение зрения, дисбаланс и когнитивные нарушения. Диагностика включает офтальмологическое обследование (визометрия, офтальмоскопия, электроретинография), отоневрологическое обследование (аудиометрия, вестибулярные тесты), генетическое обследование. Лечение направлено на коррекцию слуха (слуховые аппараты, IC), поддержание зрения (вит. А, Е, омега-3).
|
|
Дополнительные факты
Синдром Ушера (синдром Ушера) - наиболее частая причина наследственной слепоглухоты. Распространенность его среди населения оценивается в 3,2-6,2 случая на 100 тыс. Жителей (по другим данным - 1: 6000). Наибольшая заболеваемость отмечается среди евреев-ашкенази, франко-канадцев, испанских аргентинцев, финнов и «Первооткрывателем» заболевания является немецкий офтальмолог А. фон Грефе. Однако свое «название» синдром получил в честь британского офтальмолога Ашера, который в 1914 году указал на наследственную природу болезни. Молекулярно-генетические механизмы синдрома были расшифрованы в 1995 г., что открыло широкие возможности для его изучения.
Причины
В настоящее время известно более десятка генов, дефекты которых могут привести к развитию синдрома Ашера. Все эти гены, несмотря на их различную локализацию и функции, являются частью трансмембранного белкового комплекса, участвующего в движении миозина, функционировании фоторецепторов сетчатки, а также волосковых клеток улитки. Наиболее частые мутации обнаруживаются в следующих генах:
• сDH23 - «ген глухоты», кодирующий белок кадгерин-23;
• MYO7A - ген миозина VIIA;
• PCDH15 - ген протокадгерина-15;
• USH1C - ген гармонина;
• USH1G - ген анкириноподобного белка;
• USH2A - ген ушерина;
• ADGRV1 - ген адгезии G-белков;
• сLRN1 - ген кларина-1 и.
Синдром Ушера наследуется по аутосомно-рецессивному типу от обоих родителей, несущих дефектные гены. Чаще болеют представители закрытых этносов, среди которых довольно часты близкородственные браки.
• сDH23 - «ген глухоты», кодирующий белок кадгерин-23;
• MYO7A - ген миозина VIIA;
• PCDH15 - ген протокадгерина-15;
• USH1C - ген гармонина;
• USH1G - ген анкириноподобного белка;
• USH2A - ген ушерина;
• ADGRV1 - ген адгезии G-белков;
• сLRN1 - ген кларина-1 и.
Синдром Ушера наследуется по аутосомно-рецессивному типу от обоих родителей, несущих дефектные гены. Чаще болеют представители закрытых этносов, среди которых довольно часты близкородственные браки.
Патогенез
Белковые продукты, кодируемые этими генами, непосредственно участвуют в развитии и функционировании рецепторного аппарата сетчатки и внутреннего уха. Генетические мутации приводят к нарушению формирования аппарата восприятия - фоторецепторов и волосковых клеток, что сопровождается врожденными или ранними нарушениями функций зрения, слуха и равновесия.
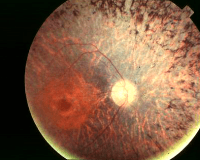
При синдроме Ашера пигментные гранулы собираются на глазном дне и распространяются от центра к периферии. Со временем происходит сужение полей и снижение остроты зрения. Патология поражает и структуры внутреннего уха: наблюдается атрофия спирального узла, нервных волокон кортиевого органа и сосудистой полоски улитки.
При синдроме Ашера пигментные гранулы собираются на глазном дне и распространяются от центра к периферии. Со временем происходит сужение полей и снижение остроты зрения. Патология поражает и структуры внутреннего уха: наблюдается атрофия спирального узла, нервных волокон кортиевого органа и сосудистой полоски улитки.
Классификация
Синдром Ушера генетически неоднороден. Выделяют 4 клинических подтипа, которые различаются молекулярно-генетическими механизмами, возрастом проявления и тяжестью симптомов:
• Тип 1. Связан с мутациями в генах MYO7A, сDH23, PCDH15, USH1G, USH1C. Курс посложнее. Характерны врожденная глухота или глубокая потеря слуха. Вестибулярные нарушения и признаки пигментного ретинита развиваются до 5 лет. На его долю приходится около 30% всех случаев.
• Тип 2. Вызван мутациями USH2A, DFNB31, ADGRV1. Сопровождается непрогрессирующей тугоухостью, нарушением зрения через 10 лет. Вестибулярная функция не нарушена. Выявляется примерно у 60% пациентов.
• Тип 3. Связан с мутациями в гене сLRN1. Он протекает с постепенным ухудшением слуха и зрения, часто с вестибулярной дисфункцией. Изменения сетчатки развиваются через 20 лет. Выявляется у 3% пациентов.
• Тип 4. Атипичный вариант синдрома Ашера. Это вызвано дефектами генов HARS, PDZD7, сEP250, с2orf71, он может быть унаследован с X-сцеплением.
• Тип 1. Связан с мутациями в генах MYO7A, сDH23, PCDH15, USH1G, USH1C. Курс посложнее. Характерны врожденная глухота или глубокая потеря слуха. Вестибулярные нарушения и признаки пигментного ретинита развиваются до 5 лет. На его долю приходится около 30% всех случаев.
• Тип 2. Вызван мутациями USH2A, DFNB31, ADGRV1. Сопровождается непрогрессирующей тугоухостью, нарушением зрения через 10 лет. Вестибулярная функция не нарушена. Выявляется примерно у 60% пациентов.
• Тип 3. Связан с мутациями в гене сLRN1. Он протекает с постепенным ухудшением слуха и зрения, часто с вестибулярной дисфункцией. Изменения сетчатки развиваются через 20 лет. Выявляется у 3% пациентов.
• Тип 4. Атипичный вариант синдрома Ашера. Это вызвано дефектами генов HARS, PDZD7, сEP250, с2orf71, он может быть унаследован с X-сцеплением.
Клиническая картина
Классическая клиническая картина развивается при первом типе синдрома Ушера. В раннем детстве у ребенка диагностируют тяжелую нейросенсорную тугоухость или полную глухоту. Отмечается задержка психомоторного развития, дети поздно начинают ходить. В дошкольном возрасте выявляется нарушение функции зрения: в результате обострения пигментного ретинита быстро ухудшается периферическое зрение и развивается куриная слепота (гемералопия). Это проявляется затруднением ориентации в темноте, частыми поездками, столкновениями с препятствием (другим человеком, дверью, мебелью).
Возникают вестибулярные нарушения: головокружение, нарушение равновесия, атактическая походка. В некоторых случаях могут наблюдаться когнитивные нарушения, психозы. При других типах болезни Ашера зрительные и слуховые нарушения развиваются позже и менее выражены; вестибулярный синдром не наблюдается.
Ассоциированные симптомы: Шаткая походка.
Возникают вестибулярные нарушения: головокружение, нарушение равновесия, атактическая походка. В некоторых случаях могут наблюдаться когнитивные нарушения, психозы. При других типах болезни Ашера зрительные и слуховые нарушения развиваются позже и менее выражены; вестибулярный синдром не наблюдается.
Ассоциированные симптомы: Шаткая походка.
Возможные осложнения
Нейросенсорная тугоухость и постепенная потеря зрения приводят к тяжелой инвалидности. Дети с синдромом Ашера нуждаются в специальном образовании, психологической и педагогической поддержке и создании безопасной среды. Вестибулярные расстройства могут стать причиной падений и повысить риск травм. Речевая и интеллектуальная сфера страдает вторично. Одно из самых частых осложнений - катаракта. Прогрессирующее снижение зрения приводит к тому, что к 40-50 годам пациенты с синдромом Ашера могут полностью ослепнуть. В тяжелых случаях возникает слепоглухота.
Диагностика
Поскольку нарушения при синдроме Ашера затрагивают разные анатомические и функциональные системы, диагноз должен быть поставлен с применением мультидисциплинарного подхода. Пациентам необходимы консультации отоларинголога, офтальмолога, генетика и комплексное инструментальное и лабораторное обследование:
• Офтальмологический визит. Визометрия и осмотр глазного дна не всегда выявляют первые признаки дегенерации пигмента сетчатки. В этом отношении наиболее чувствительным тестом является электроретинография, выявляющая изменения даже на доклиническом уровне. По результатам периметрии отмечается концентрическое сужение полей зрения.
• Исследование слуха. Степень потери слуха определяется с помощью аудиометрии. Кроме того, записывается отоакустическая эмиссия, ВП ствола, проводится электрокхлеарение. Больной осмотрен сурдологом.
• Исследование вестибулярного анализатора. Для выявления вестибулярных нарушений проводится видеонистагмография, вестибулометрические пробы.
• Генодиагностика. Выполняется секвенирование образцов генетического материала пациента. На основании обнаруженных изменений определяется генетический вариант синдрома Ашера. Также требуется медико-генетическое консультирование членов семьи.
• Офтальмологический визит. Визометрия и осмотр глазного дна не всегда выявляют первые признаки дегенерации пигмента сетчатки. В этом отношении наиболее чувствительным тестом является электроретинография, выявляющая изменения даже на доклиническом уровне. По результатам периметрии отмечается концентрическое сужение полей зрения.
• Исследование слуха. Степень потери слуха определяется с помощью аудиометрии. Кроме того, записывается отоакустическая эмиссия, ВП ствола, проводится электрокхлеарение. Больной осмотрен сурдологом.
• Исследование вестибулярного анализатора. Для выявления вестибулярных нарушений проводится видеонистагмография, вестибулометрические пробы.
• Генодиагностика. Выполняется секвенирование образцов генетического материала пациента. На основании обнаруженных изменений определяется генетический вариант синдрома Ашера. Также требуется медико-генетическое консультирование членов семьи.
|
|
Диф. диагностика
Синдром Ушера необходимо отличать от других синдромных и несиндромальных форм глухоты и слепоты, которые развиваются при:
• Врожденная краснуха - врожденная потеря слуха, катаракта, врожденные пороки сердца;
• Синдром Альстрома - дегенерация сетчатки, нейросенсорная тугоухость, ожирение, сахарный диабет, кардиомиопатия, нефропатия;
• Синдром Халльгрена (акусто-ретино-мозжечковая дегенерация) - пигментный ретинит, катаракта, потеря слуха, мозжечковый синдром.
• Врожденная краснуха - врожденная потеря слуха, катаракта, врожденные пороки сердца;
• Синдром Альстрома - дегенерация сетчатки, нейросенсорная тугоухость, ожирение, сахарный диабет, кардиомиопатия, нефропатия;
• Синдром Халльгрена (акусто-ретино-мозжечковая дегенерация) - пигментный ретинит, катаракта, потеря слуха, мозжечковый синдром.
Лечение
На сегодняшний день не разработаны методы полного излечения от болезни. Все принимаемые меры направлены на компенсацию нарушенных функций и замедление течения болезни. Пациентам с синдромом Ашера рекомендуется ежегодно проходить поддерживающие курсы медикаментозной терапии, включая ноотропы, вазодилататоры и антиоксиданты. Чтобы подавить прогрессирование пигментного ретинита, рекомендуется принимать высокие дозы пальмитата ретинола и поддерживать диету, богатую витаминами А, Е и ПНЖК.
Способ коррекции функции слуха подбирается индивидуально. Выбор может быть сделан в пользу слуховых аппаратов или кохлеарных имплантатов. Для адаптации детей с синдромом Ашера к жизни в обществе важна помощь психологов и глухих учителей.
Сообщается о разработке генной терапии для пациентов с синдромом Ушера 1 типа. UshStat, лентивирусный вектор для доставки интактного гена MYO7A, в настоящее время проходит клинические испытания. Для восстановления зрительной функции предлагается субретинальное введение УшСтата.
Способ коррекции функции слуха подбирается индивидуально. Выбор может быть сделан в пользу слуховых аппаратов или кохлеарных имплантатов. Для адаптации детей с синдромом Ашера к жизни в обществе важна помощь психологов и глухих учителей.
Сообщается о разработке генной терапии для пациентов с синдромом Ушера 1 типа. UshStat, лентивирусный вектор для доставки интактного гена MYO7A, в настоящее время проходит клинические испытания. Для восстановления зрительной функции предлагается субретинальное введение УшСтата.
Прогноз
На качество жизни пациентов с синдромом Ашера в значительной степени влияют проблемы со зрением и слухом. Около 50% людей с типом 1 и 70% с заболеванием типа 2 поддерживают от 20 до 40% остроты зрения на оба или один глаз. Пациентам необходима пожизненная диета, защита глаз от ультрафиолета. Профилактика заключается в генетическом консультировании и лабораторных исследованиях в семьях с заболевшими. Возможный пренатальный диагноз синдрома Ушера.
Список литературы
1. Изучение интерактома при синдроме Ашера в российской популяции для выбора приоритетных патогенетически ориентированных терапевтических подходов/ Иванова М.Е , Атарщиков Д.С. Демчинский А.М. Стрельников В.В. Бар Д. Порядин Г.В. Балашова Л.М. Салмаси Ж.М. РМЖ «Клиническая Офтальмология». 2019. №4.
2. Сравнительная оценка заболеваемости синдромом Ушера в.
3. Этиологические аспекты врожденной тугоухости/ Коноплев О.И. и / Медицинский вестник Северного Кавказа. 2019.
2. Сравнительная оценка заболеваемости синдромом Ушера в.
3. Этиологические аспекты врожденной тугоухости/ Коноплев О.И. и / Медицинский вестник Северного Кавказа. 2019.