Другие названия и синонимы
Lawrence-Moon-Bardet-Bidl syndrome.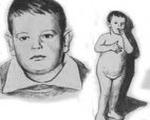
МКБ-10 коды
|
|
Описание
Синдром Лорана-Люна-Барде-Бидля. Это аутосомно-рецессивное нейроэндокринное заболевание, которое характеризуется ожирением, дегенеративными поражениями сетчатки, интеллектуальными нарушениями. Патология также проявляется разнообразными деформациями скелета, врожденными пороками развития внутренних органов и неврологическими осложнениями. Для диагностики заболевания необходимы нейровизуализация, исследование глазного дна, комплексное лабораторное обследование. Поддерживающая терапия включает прием лекарств (ноотропы, гормоны), диету, логопедию и коррекцию дефектов.
Дополнительные факты
Первые сведения о патологии появились в 1866 году, когда английский офтальмолог Д. Лоуренс и американский офтальмолог Р. Мун описали семейный случай ожирения, умственной отсталости и впоследствии развился тетрапарез. В 1920-е гг. В двадцатом веке французский врач Ж. Барде и венгерский специалист А. Бидль объявили об аналогичных клинических наблюдениях, которые в 1925 г. были объединены в одно заболевание - синдром Лоуренса-Муна-Барде-Бидля. В Европе и США заболевание встречается с частотой 1 случай на 140-160 тысяч жителей, а в странах Азии и Африки - в 2-3 раза чаще.
Причины
На сегодняшний день описано более 15 вариантов дефектов генов, при которых встречается синдром Лоуренса-Муна-Барде-Бидля. 45% случаев связаны с мутацией в локусе вBS1 (11q13), также часто встречаются изменения на вBS2 (16q22), вBS3 (3p13), вBS4 (15q21). Заболевание передается по аутосомно-рецессивному типу, однако явный семейный характер можно проследить только у 40% пациентов. Патология имеет переменное проникновение.
Патогенез
По механизму развития синдром относится к категории цилиопатий, для которых характерно нарушение строения и функционирования ресничек. В норме эти образования располагаются на поверхности клеток различных тканей, и делятся на первичные (неподвижные) и вторичные (подвижные). Для патологии Лоуренса-Муна-Барде-Бидля типичное поражение первичных ресничек, состоящее из 9 пар периферических микротрубочек, одной пары центральных микротрубочек.
Несмотря на свою неподвижность, реснички участвуют во многих физиологических процессах в клетке: они помогают в транспортировке клеточных сигналов, влияют на характеристики протекания биохимических реакций и способствуют правильной ориентации плоскости деления. В физиологических условиях они содержатся в фоторецепторах, киноцилиях, остеоцитах и других клетках внутренних органов, поэтому симптоматический полиморфизм наблюдается у страдающих синдромом Лоуренса-Муна-Барде-Бидля.
Несмотря на свою неподвижность, реснички участвуют во многих физиологических процессах в клетке: они помогают в транспортировке клеточных сигналов, влияют на характеристики протекания биохимических реакций и способствуют правильной ориентации плоскости деления. В физиологических условиях они содержатся в фоторецепторах, киноцилиях, остеоцитах и других клетках внутренних органов, поэтому симптоматический полиморфизм наблюдается у страдающих синдромом Лоуренса-Муна-Барде-Бидля.
Клиническая картина
Первые клинические проявления синдрома появляются с раннего детства, но из-за разнообразия симптомов и отсутствия патогномоничных признаков не всегда своевременно диагностируются. Наиболее типичные органы-мишени: глаза, длинные кости, органы эндокринной системы, головной мозг. Некоторые ученые предлагают разделять синдромы Лоуренса-Муна и Барда-Бидля по отдельности, но эта теория пока не получила должной поддержки.
Избыточный вес характерен для синдрома Лоуренса-Муна-Барде-Бидля, у 72-92% пациентов ожирение формируется по центральному типу - максимальное количество жировых отложений сосредоточено в области живота. У половины пациентов сахарный диабет 2 типа развивается в результате нарушения обмена веществ. Тяжелый гипогонадизм (недоразвитие половых органов) встречается у 74-85% мужчин и 45-53% женщин.
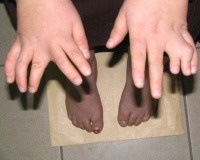
Специфическими симптомами заболевания являются пороки развития скелета коротких пальцев (брахидактилия), встречающиеся у 42-100% пациентов, увеличение количества пальцев рук или ног (полидактилия) - 63-81%, анастомоз пальцев рук ( синдактилия) - 8-95%. Повреждение зрительного аппарата представляет собой в основном пигментный ретинит (у 93% пациентов), приводящий к прогрессирующему ухудшению зрения. Реже диагностируют астигматизм, косоглазие, катаракту.
Другой распространенной группой симптомов при болезни Лоуренса-Муна-Барде-Бидля являются нервно-психические расстройства. В раннем детстве у 90% пациентов наблюдается задержка речевого развития разной степени выраженности, а затем более 80% пациентов имеют дефекты речи. Умственная отсталость встречается в 75-90% случаев. Неврологические расстройства представлены нарушением координации движений, снижением функции обонятельного нерва.
Ассоциированные симптомы: Ограниченный словарный запас. Отсутствие речи.
Избыточный вес характерен для синдрома Лоуренса-Муна-Барде-Бидля, у 72-92% пациентов ожирение формируется по центральному типу - максимальное количество жировых отложений сосредоточено в области живота. У половины пациентов сахарный диабет 2 типа развивается в результате нарушения обмена веществ. Тяжелый гипогонадизм (недоразвитие половых органов) встречается у 74-85% мужчин и 45-53% женщин.
Специфическими симптомами заболевания являются пороки развития скелета коротких пальцев (брахидактилия), встречающиеся у 42-100% пациентов, увеличение количества пальцев рук или ног (полидактилия) - 63-81%, анастомоз пальцев рук ( синдактилия) - 8-95%. Повреждение зрительного аппарата представляет собой в основном пигментный ретинит (у 93% пациентов), приводящий к прогрессирующему ухудшению зрения. Реже диагностируют астигматизм, косоглазие, катаракту.
Другой распространенной группой симптомов при болезни Лоуренса-Муна-Барде-Бидля являются нервно-психические расстройства. В раннем детстве у 90% пациентов наблюдается задержка речевого развития разной степени выраженности, а затем более 80% пациентов имеют дефекты речи. Умственная отсталость встречается в 75-90% случаев. Неврологические расстройства представлены нарушением координации движений, снижением функции обонятельного нерва.
Ассоциированные симптомы: Ограниченный словарный запас. Отсутствие речи.
Возможные осложнения
Течение синдрома Лоуренса-Люна-Барде-Бидля часто осложняется множеством соматических заболеваний. В 68% случаев почечная недостаточность развивается из-за гидронефроза, поликистозных аномалий или других аномалий почек. Более половины пациентов сталкиваются с проблемами зубов, обширным кариесом. К более редким осложнениям наследственных заболеваний относятся фиброз печени, врожденные пороки сердца.
Для патологии характерно быстрое прибавление в весе, из-за чего ожирение наблюдается с дошкольного возраста и быстро достигает 3-4 степени. На этом фоне возникают серьезные нарушения обмена веществ, значительно возрастает риск эндокринных заболеваний, сердечно-сосудистых заболеваний - у 50% пациентов старше 34 лет проявляется артериальная гипертензия. При тяжелом поражении структур мозга, судорогах, спастической параплегии или квадриплегии возможны экстрапирамидные нарушения.
Для патологии характерно быстрое прибавление в весе, из-за чего ожирение наблюдается с дошкольного возраста и быстро достигает 3-4 степени. На этом фоне возникают серьезные нарушения обмена веществ, значительно возрастает риск эндокринных заболеваний, сердечно-сосудистых заболеваний - у 50% пациентов старше 34 лет проявляется артериальная гипертензия. При тяжелом поражении структур мозга, судорогах, спастической параплегии или квадриплегии возможны экстрапирамидные нарушения.
Диагностика
При подробной клинической картине поставить диагноз не сложно, диагноз синдрома Лоуренса-Муна-Барде-Бидля возможен после клинического осмотра педиатром или генетиком. Однако педиатры часто не обращают внимания на незавершенные формы заболевания, поэтому поддерживающее лечение назначается поздно. Для подтверждения диагноза используются следующие методы:
• Рентген скелета. Обследование проводится с целью детального изучения строения пальцев рук и ног, оценки степени деформации костей, подготовки к хирургической коррекции полидактилии и синдактилии.
• Офтальмоскопия. По всей поверхности глазного дна определяется скопление пигмента одновременно с сужением артерий сетчатки, признаками дегенерации в макулярной зоне. Это изображение указывает на наличие пигментного ретинита.
• Нейровизуализация. По результатам КТ или МРТ головного мозга у большинства пациентов выявляется агенезия мозолистого тела, аномалии структур задней черепной ямки. У пациентов с судорожным синдромом диагностика завершается с помощью ЭЭГ.
• Обследование почек. Как метод скрининговой диагностики используется УЗИ почек, по результатам которого выявляются врожденные аномалии строения органа. Функциональную активность почек оценивают с помощью сцинтиграфии почек.
• Лабораторные методы. Пациентам рекомендуется пройти обширный биохимический анализ крови, липидный профиль, тест на сахар в крови натощак и тест на толерантность к глюкозе. Кроме того, изучаются уровни инсулина, С-пептида и половых гормонов.
• Рентген скелета. Обследование проводится с целью детального изучения строения пальцев рук и ног, оценки степени деформации костей, подготовки к хирургической коррекции полидактилии и синдактилии.
• Офтальмоскопия. По всей поверхности глазного дна определяется скопление пигмента одновременно с сужением артерий сетчатки, признаками дегенерации в макулярной зоне. Это изображение указывает на наличие пигментного ретинита.
• Нейровизуализация. По результатам КТ или МРТ головного мозга у большинства пациентов выявляется агенезия мозолистого тела, аномалии структур задней черепной ямки. У пациентов с судорожным синдромом диагностика завершается с помощью ЭЭГ.
• Обследование почек. Как метод скрининговой диагностики используется УЗИ почек, по результатам которого выявляются врожденные аномалии строения органа. Функциональную активность почек оценивают с помощью сцинтиграфии почек.
• Лабораторные методы. Пациентам рекомендуется пройти обширный биохимический анализ крови, липидный профиль, тест на сахар в крови натощак и тест на толерантность к глюкозе. Кроме того, изучаются уровни инсулина, С-пептида и половых гормонов.
Лечение
Специфического лечения заболевания не существует. Медицинскую помощь оказывает многопрофильная команда, состоящая из эндокринолога, невролога, нейропсихолога, тюремных учителей и других специалистов. С учетом ведущих клинических симптомов у данного пациента используются следующие направления терапии:
• Диетотерапия. В целях контроля массы тела, предотвращения сахарного диабета и гиперлипидемии выбирается диета с пониженным количеством простых углеводов и насыщенных жиров. Пациентам рекомендуется сбалансированная диета с повышенным содержанием витаминов и микроэлементов.
• Гормональная терапия. Заместительная терапия гонадотропинами применяется для коррекции гипогонадизма, возможного восстановления репродуктивной функции. В случае нарушения обмена веществ может потребоваться инсулин.
• Защита глаз от ультрафиолета. Для профилактики пигментного ретинита пациентам показывают, как носить качественные солнцезащитные очки, в подростковом возрасте рассматривается вариант контактных линз с УФ-фильтрами.
• Ноотропная терапия. Назначаются определенные препараты, стимулирующие мозговое кровообращение, улучшающие обменные процессы в нервной ткани. Лечение позволяет скорректировать умственную отсталость, задержку развития речевых навыков.
• Нейропсихологическая реабилитация. Пациентам с пониженным интеллектом требуется помощь дефектологов и подбор специальных программ обучения. Для устранения нарушений речи необходима логопедическая коррекция.
• Эксплуатация. Помощь хирургов необходима для устранения врожденных аномалий пальцев рук и ног, улучшения их функциональных возможностей, достижения максимально возможного косметического эффекта.
Учитывая возможные осложнения, пациентам с синдромом Лоуренса-Муна-Барде-Бидля требуется пожизненное амбулаторное наблюдение у офтальмолога, кардиолога и нефролога. С целью контроля состояния внутренних органов регулярно проводятся лабораторные и инструментальные исследования, при обнаружении отклонений в показателях здоровья назначается соответствующее лечение.
• Диетотерапия. В целях контроля массы тела, предотвращения сахарного диабета и гиперлипидемии выбирается диета с пониженным количеством простых углеводов и насыщенных жиров. Пациентам рекомендуется сбалансированная диета с повышенным содержанием витаминов и микроэлементов.
• Гормональная терапия. Заместительная терапия гонадотропинами применяется для коррекции гипогонадизма, возможного восстановления репродуктивной функции. В случае нарушения обмена веществ может потребоваться инсулин.
• Защита глаз от ультрафиолета. Для профилактики пигментного ретинита пациентам показывают, как носить качественные солнцезащитные очки, в подростковом возрасте рассматривается вариант контактных линз с УФ-фильтрами.
• Ноотропная терапия. Назначаются определенные препараты, стимулирующие мозговое кровообращение, улучшающие обменные процессы в нервной ткани. Лечение позволяет скорректировать умственную отсталость, задержку развития речевых навыков.
• Нейропсихологическая реабилитация. Пациентам с пониженным интеллектом требуется помощь дефектологов и подбор специальных программ обучения. Для устранения нарушений речи необходима логопедическая коррекция.
• Эксплуатация. Помощь хирургов необходима для устранения врожденных аномалий пальцев рук и ног, улучшения их функциональных возможностей, достижения максимально возможного косметического эффекта.
Учитывая возможные осложнения, пациентам с синдромом Лоуренса-Муна-Барде-Бидля требуется пожизненное амбулаторное наблюдение у офтальмолога, кардиолога и нефролога. С целью контроля состояния внутренних органов регулярно проводятся лабораторные и инструментальные исследования, при обнаружении отклонений в показателях здоровья назначается соответствующее лечение.
|
|
Прогноз
Синдром Лоуренса-Муна-Барде-Бидля относится к неизлечимым патологиям, но при раннем начале терапии можно значительно улучшить качество жизни пациентов, предотвратить опасные для жизни осложнения. Менее оптимистичный прогноз при наличии тяжелых врожденных пороков развития в результате полиорганной недостаточности. В целях профилактики заболеваний парам с отягощенной наследственностью показаны медицинские и генетические консультации по вопросам планирования семьи.
Список литературы
1. Синдром Барде-Бидля/ Е.А. Потрохова, М.Л. Баабаян, Л.С. Балева// Российский вестник перинатологии и педиатрии. 2020. №6.
2. Синдром Барде-Бидля с врожденной аномалией развития почек у девочки 14 лет/ А.В. Бурлуцкая, Н.В. Савельева// Кубанский научный медицинкий вестник. 2019. №3.
3. Синдром Лоренса-Муна-Барде-Бидля/ С.Д. Касымова, М.А. Мирахмедова// Вестник последипломного образования в сфере здравоохранения. 2017. №3.
4. Ранняя диагностика синдрома Барде-Бидля, ассоциированного с ожирением/ Н.Н. Волеводз, И.А. Еремина, Т.В. Семичева// Ожирение и метаболизм. 2008. №1.
2. Синдром Барде-Бидля с врожденной аномалией развития почек у девочки 14 лет/ А.В. Бурлуцкая, Н.В. Савельева// Кубанский научный медицинкий вестник. 2019. №3.
3. Синдром Лоренса-Муна-Барде-Бидля/ С.Д. Касымова, М.А. Мирахмедова// Вестник последипломного образования в сфере здравоохранения. 2017. №3.
4. Ранняя диагностика синдрома Барде-Бидля, ассоциированного с ожирением/ Н.Н. Волеводз, И.А. Еремина, Т.В. Семичева// Ожирение и метаболизм. 2008. №1.