Другие названия и синонимы
Chediak-Higashi syndrome.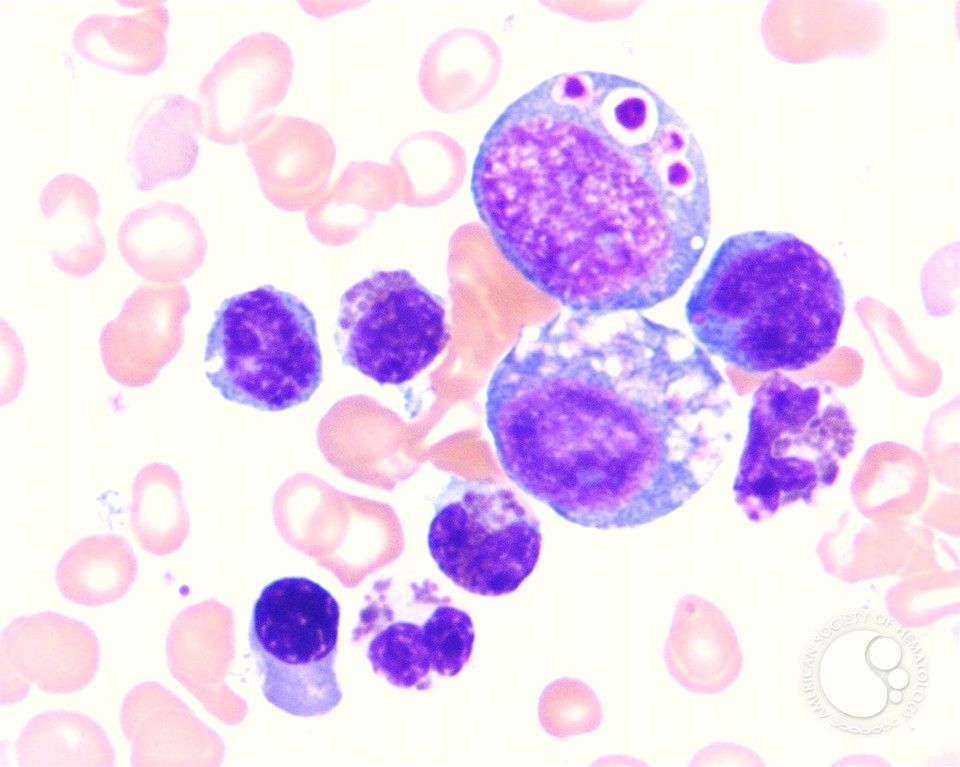
МКБ-10 коды
- МКБ-10
- D72.1 Эозинофилия
|
|
Описание
Синдром Чедиака-Хигаси (по-английски сhediak-Higashi syndrome, сHS) - заболевание с генерализованной клеточной дисфункцией. Тип наследования - аутосомно-рецессивный. Обусловлено дефектом Lyst-протеина. Характерный признак этого синдрома - гигантские пероксидазоположительные гранулы в нейтрофилах, эозинофилах, моноцитах периферической крови и костного мозга, а также в клетках-предшественницах гранулоцитов. Гигантские гранулы обнаруживают также в циркулирующих лимфоцитах, цитоплазме нейронов и клетках соединительной ткани периневральной области.
Синдром Чедиака-Хигаши - редкое заболевание, характеризующееся тяжелыми повторными гнойными инфекциями, парциальным альбинизмом, прогрессирующей нейропатией, склонностью к кровотечениям, развитием лимфопролиферативного синдрома, а также наличием гигантских гранул во многих клетках, особенно в лейкоцитах периферической крови. Иммунодефицит при синдроме Чедиака-Хигаши обусловлен, в первую очередь, нарушением функции фагоцитоза в клетках гранулоцитарного и макрофагального ряда и проявляется склонностью к гнойным и грибковым инфекциям. Кровоточивость связана с дефектом высвобождения фомбоцитаркых гранул.
Первое упоминание о синдроме Чедиака-Хигаши относится к 1943 году (Beguez сesar). Дальнейшие описания мы находим у Steinbrinck 1948, сhediak 1952 и, наконец, у Higashi 1954.
Синдром Чедиака-Хигаши - редкое заболевание, характеризующееся тяжелыми повторными гнойными инфекциями, парциальным альбинизмом, прогрессирующей нейропатией, склонностью к кровотечениям, развитием лимфопролиферативного синдрома, а также наличием гигантских гранул во многих клетках, особенно в лейкоцитах периферической крови. Иммунодефицит при синдроме Чедиака-Хигаши обусловлен, в первую очередь, нарушением функции фагоцитоза в клетках гранулоцитарного и макрофагального ряда и проявляется склонностью к гнойным и грибковым инфекциям. Кровоточивость связана с дефектом высвобождения фомбоцитаркых гранул.
Первое упоминание о синдроме Чедиака-Хигаши относится к 1943 году (Beguez сesar). Дальнейшие описания мы находим у Steinbrinck 1948, сhediak 1952 и, наконец, у Higashi 1954.
Клиническая картина
Клинические проявления синдрома Чедиака-Хигаси - рецидивирующие пиогенные инфекции, характерны частичный альбинизм волос, кожи и глаз, фотофобия. Вскоре после рождения возникает торпидная фаза болезни, связанная с аномалией образования антител к вирусу Эпстайна-Барр. Клинически на фоне бактериальной или вирусной инфекции развивается вторичный гемофагоцитарный синдром; лихорадка, панцитопения с геморрагическим синдромом, лимфаденопатия, гепатоспленомегалия, неврологическая симптоматика - эпизоды судорог, нарушение чувствительности, парезы, мозжечковые нарушения, умственная отсталость. Прогноз неблагоприятен.
Причины
Тип наследования - аутосомно-рецессивный. Обусловлено дефектом Lyst-протеина.
Патогенез
Патогенез заболевания связывают с аномалией структуры клеточных мембран, нарушением системы собирательных микротрубочек и дефектом взаимодействия последних с мембранами лизосом. Большая часть клинических проявлений может быть объяснена аномальным распределением лизосомальных ферментов. Частота и тяжесть пиогенных инфекций обусловлена снижением активности кислородного метаболизма и внутриклеточного переваривания микробов в фагоцитах вследствие задержки и непостоянного высвобождения гидролитических лизосомальных ферментов из гигантских гранул в фагосомы. Кроме того, у больных снижены активность естественных киллеров и антителозависимая цитотоксичность лимфоцитов. Заболевание относят к первичным иммунодефицитам.
Лечение
При лечении синдрома Чедиака-Хигаси принимаются симптоматические меры, защита кожи и глаз от инсоляции. В лечении инфекционных эпизодов - комбинация антибиотиков широкого спектра действия. При развитии гемофагоцитоза показана полихимиотерапия с включением глюкокортикосероидов (преимущественно дексаметазона), винкристина, этопозида, эндолюмбальными введениями метотрексата, заместительной терапией компонентами крови. Единственный радикальный метод лечения, как и при многих других первичных иммунодефицитах, - аллогенная трансплантация костного мозга.