МКБ-10 коды
|
|
Вступление
Кодирование по Международной статистической классификации болезней и проблем, связанных со здоровьем: E76.0.
Год утверждения (частота пересмотра): 2019.
Возрастная категория: Взрослые.
Пересмотр не позднее: 2021.
ID: 291.
Разработчик клинической рекомендации.
• Ассоциация медицинских генетиков.
• Союз педиатров России.
Одобрено Научно-практическим Советом Минздрава РФ.
Год утверждения (частота пересмотра): 2019.
Возрастная категория: Взрослые.
Пересмотр не позднее: 2021.
ID: 291.
Разработчик клинической рекомендации.
• Ассоциация медицинских генетиков.
• Союз педиатров России.
Одобрено Научно-практическим Советом Минздрава РФ.
|
|
 Список сокращений
Список сокращений
АЛТ - аланинаминотрансфераза;
АСТ - аспартатаминотрансфераза;
ГАГ - гликозаминогликаны;
КТ - компьютерная томография;
МПС - мукополисахаридоз;
МРТ - магнитно-резонансная томография;
ССС - сердечно - сосудистая система;
ТГСК - трансплантации гемопоэтических стволовых клеток;
УЗИ - ультразвуковое исследование;
ФВД - функция внешнего дыхания;
ФЗТ - ферментная заместительная терапия;
ЭКГ - электрокардиография;
ЭНМГ - электронейромиография;
Эхо-КГ - эхокардиография;
ЭЭГ - электроэнцефалография.
АСТ - аспартатаминотрансфераза;
ГАГ - гликозаминогликаны;
КТ - компьютерная томография;
МПС - мукополисахаридоз;
МРТ - магнитно-резонансная томография;
ССС - сердечно - сосудистая система;
ТГСК - трансплантации гемопоэтических стволовых клеток;
УЗИ - ультразвуковое исследование;
ФВД - функция внешнего дыхания;
ФЗТ - ферментная заместительная терапия;
ЭКГ - электрокардиография;
ЭНМГ - электронейромиография;
Эхо-КГ - эхокардиография;
ЭЭГ - электроэнцефалография.
Термины и определения
Ферментная заместительная терапия - лечение, заключающееся во введении препарата (рекомбинантного фермента) пациентам с наследственным нарушением метаболизма.
Лизосомные болезни накопления - группа наследственных моногенных заболеваний, связанных с нарушением функции лизосом.
Мукополисахаридозы (МПС) - группа наследственных болезней обмена веществ, связанных с нарушением метаболизма гликозаминогликанов (ГАГ), приводящее к поражению органов и тканей. Обусловлены данные заболевания мутациями генов, контролирующих процесс внутрилизосомного гидролиза макромолекул.
Лизосомные болезни накопления - группа наследственных моногенных заболеваний, связанных с нарушением функции лизосом.
Мукополисахаридозы (МПС) - группа наследственных болезней обмена веществ, связанных с нарушением метаболизма гликозаминогликанов (ГАГ), приводящее к поражению органов и тканей. Обусловлены данные заболевания мутациями генов, контролирующих процесс внутрилизосомного гидролиза макромолекул.
Описание
Мукополисахаридоз I типа (МПС I) - наследственная лизосомная болезнь накопления, обусловленная дефицитом фермента альфа-L-идуронидазы и протекающая с различными клиническими проявлениями: задержкой роста, умственной отсталостью, поражением нервной системы, сердечно-легочными нарушениями, гепатоспленомегалией, множественными дизостозами, помутнением роговицы. Все вышеперечисленные признаки приводят к инвалидизации, а при тяжелом течении болезни - к летальному исходу [1].
Причины
Причиной МПС I типа является мутация в гене, кодирующем лизосомный фермент альфа-L-идуронидазу. Тип наследования: аутосомно-рецессивный. Ген IDUA, кодирующий альфа-L-идуронидазу локализован в хромосомной области 4p16,3.
Из-за снижения активности фермента происходит накопление различных типов ГАГ и развивается соматическая манифестация в виде лицевого дисморфизма, гепатоспленомегалии, поражения сердца, дыхательной системы, изменений скелета, неврологической симптоматики, гематологические и офтальмологические изменения. Вариабельность МПС определяется типом накапливаемого субстрата при недостаточной деградации ГАГ: при МПС I типа происходит накопление гепарансульфата и дерматансульфата. Дефицит альфа-L-идуронидазы может привести к развитию различных фенотипов болезни, обусловливая отличия в тяжести симптоматики. Выделяют три клинических фенотипа [3,4,5]:
-синдром Гурлер (мукополисахаридоз I H - тяжелая форма),.
-синдром Шейе (мукополисахаридоз I S - легкая форма).
-синдром Гурлер-Шейе (мукополисахаридоз I H/S-промежуточная форма).
Из-за снижения активности фермента происходит накопление различных типов ГАГ и развивается соматическая манифестация в виде лицевого дисморфизма, гепатоспленомегалии, поражения сердца, дыхательной системы, изменений скелета, неврологической симптоматики, гематологические и офтальмологические изменения. Вариабельность МПС определяется типом накапливаемого субстрата при недостаточной деградации ГАГ: при МПС I типа происходит накопление гепарансульфата и дерматансульфата. Дефицит альфа-L-идуронидазы может привести к развитию различных фенотипов болезни, обусловливая отличия в тяжести симптоматики. Выделяют три клинических фенотипа [3,4,5]:
-синдром Гурлер (мукополисахаридоз I H - тяжелая форма),.
-синдром Шейе (мукополисахаридоз I S - легкая форма).
-синдром Гурлер-Шейе (мукополисахаридоз I H/S-промежуточная форма).
Эпидемиология
Мукополисахаридоз I типа - это панэтническое заболевание, его частота составляет 1:100 000 новорожденных. Приблизительно 50%-80% пациентов имеют тяжелую форму заболевания. МПС I H/S синдром Гурлер-Шейе встречается с популяционной частотой 1:100 000 - 1:500 000 новорожденных; МПС I S синдром Шейе - 1:500 000 новорожденных [6,7]. Однако, нужно учитывать, что существует определенная погрешность в оценке распространенности различных фенотипов заболевания, что может быть связано с более частым выявлением именно тяжелых форм МПС I.
E76,0. Мукополисахаридоз I типа.
1,4 Особенности кодирования заболевания или состояния (группы заболеваний или состояний) по Международной статистической классификации болезней и проблем, связанных со здоровьем.
Согласно МКБ10, заболевание относится к классу IV, болезням эндокринной системы, расстройству питания и нарушению обмена веществ,.E76,0. Мукополисахаридоз I типа.
Классификация
1,5 Классификация заболевания или состояния (группы заболеваний или состояний).
В соответствии с дефицитом/отсутствием лизосомных ферментов, соответствующим генным дефектам и тяжести клинической симптоматики, выделяют 11 типов МПС (приложение Г1).Клиническая картина
В настоящее время МПС I рассматривается как заболевание с континуумом клинических фенотипов, различающихся по возрасту манифестации, тяжести клинических проявлений и скорости прогрессирования заболевания у пациентов [1-5].
Таким образом, довольно условно выделяют тяжелую форму заболевания (синдром Гурлер), с манифестацией на первом году жизни, с прогрессирующей кардио-респираторной недостаточностью и ярко выраженной неврологической симптоматикой; и мягкую форму (синдромы Гурлер-Шейе и Шейе), при которой симптомы появляются в возрасте 4-10 лет и болезнь медленно прогрессирует, при этом некоторые пациенты доживают до взрослого возраста.
В данных клинических рекомендациях не будет рассматриваться синдром Гурлер, так как он подробно рассмотрен в клинических рекомендациях по МПС I типа у детей и не встречается у взрослых.
С учетом традиционно используемой классификацией, ниже приведены характерные клинические признаки для МПС I различных форм.
Мукополисахаридоз тип I S - легкая форма.
Синонимы: синдром Шейе, МПС I S - легкая форма.
Основные клинические проявления: огрубление черт лица, тугоподвижность суставов, множественный дизостоз, помутнение роговицы [3,4].
Внешний вид. Первые признаки заболевания появляются в возрасте 3-5 лет. Пациенты гиперстенического телосложения с сильно развитой мускулатурой, черты лица грубые, характерен широкий рот с пухлыми губами, нижняя прогнатия. Постепенно развивается ограничение движений в суставах верхних конечностей, а также тугоподвижность, возникшие и прогрессирующие контрактуры суставов кистей. Наиболее выраженными все симптомы болезни становятся к периоду полового созревания. Возможно повышенное оволосение, короткая шея.
Костная система: может отмечается небольшая задержка роста. Множественный дизостоз: дисплазия лицевого черепа; плоская переносица, широко расставленные глаза, утолщенные губы, гипоплазированная нижняя челюсть, макроглоссия и гиперплазия десен, короткая шея, сгибательные контрактуры и тугоподвижность конечностей, с возрастом присоединяется болезненность суставов рук и стоп, с формированием «когтистой лапы» и полой стопы, вальгусной деформацией коленных суставов. Нередко развивается туннельный синдром карпального канала, который, наряду с тугоподвижностью, приводит к ограничению функции верхних конечностей. Редко - врожденный щелкающий 1 палец (болезнь Нотта).
Органы дыхания: частые респираторные заболевания в виде ринитов, отитов. Возможно развитие обструктивных заболеваний дыхательных путей, ночные апноэ.
Орган зрения: отмечается раннее неравномерное помутнение роговицы. В дальнейшем, обычно после 30 лет, развивается глаукома, пигментная дистрофия сетчатки. Редко - отек диска зрительного нерва.
Центральная нервная система: психомоторное развитие нормальное или слегка замедленное.
Краниовертебральный стеноз развивается у пациентов с МПС вследствие гипоплазии зубовидного отростка С2 позвонка, атлантоаксиальной нестабильности, отложения мукополисахаридов в твердой мозговой оболочке и задней продольной связке, что в дальнейшем приводит к компрессионной миелопатии на этом уровне, и как следствие, развитию бульбарных нарушений, центральной дыхательной недостаточности. Симптомы могут включать нарушение походки, мышечную слабость, неуклюжесть при сохранных моторных навыках и дисфункцию мочевого пузыря.
Карпальный тоннельный синдром - частая нейропатия сдавления у пациентов в возрасте от 5 до 10 лет и взрослых. При отсутствии лечения может привести к необратимой контрактуре дистальных межфаланговых суставов, а также к нарушению или потере чувствительности первых трех пальцев и парезу мышц тенара. К сожалению, пациенты редко сообщают о болевых ощущениях, пока не происходит потеря функции.
Сердечно-сосудистая система: характерны пороки аортального клапана, коарктация аорты, митральный стеноз.
Желудочно-кишечная система: отмечаются пахово-мошоночные и пупочные грыжи, нечасто - гепатоспленомегалия.
Мукополисахаридоз тип I H/S - промежуточная форма.
Синонимы: Синдром Гурлер-Шейе.
Основные клинические признаки: лицевые дисморфии, тугоподвижность суставов, низкорослость, помутнение роговицы [1, 2, 5,7].
Внешний вид: первые симптомы болезни появляются к возрасту 3-8 лет. Характерны: скафоцефалия, макроцефалия, запавшая переносица, пухлые губы, помутнение роговицы, микрогнатия, умеренный гипертрихоз, утолщение кожных покровов.
Костная система: на первом году жизни рост в пределах нормы, затем темпы роста снижаются, обусловливая низкорослость. Телосложение диспропорциональное. Множественный дизостоз, скафоцефалия, макроцефалия. Вальгусные деформации голеней незначительно выражены. Умеренно ограничена подвижность в суставах, определяются дизостозы, бочкообразная грудная клетка, кифосколиоз, гиперлордоз.
Органы дыхания: частые респираторные заболевания в виде ринитов, отитов, гипертрофия небных миндалин. Возможно развитие обструкций дыхательных путей, стеноз гортани.
Орган зрения: помутнение роговицы.
Центральная нервная система: отмечается задержка темпов психоречевого развития, позже появляется глубокая деменция. Краниовертебральный стеноз и миелопатия развивается у пациентов с МПС вследствие гипоплазии зубовидного отростка С2 позвонка, атлантоаксиальной нестабильности, отложения ГАГ в твердой мозговой оболочке и задней продольной связке, что в дальнейшем приводит к компрессионной миелопатии на этом уровне, и как следствие, развитию бульбарных нарушений, центральной дыхательной недостаточности. Симптомы могут включать нарушение походки, мышечную слабость, неуклюжесть при сохранных моторных навыках и дисфункцию мочевого пузыря. Туннельный синдром - синдром запястного канала, сообщающаяся гидроцефалия. Характерно развитие пахименингита в шейном отделе, приводящего к сдавлению спинного мозга и последующей миелопатии.
Сердечно-сосудистая система: клапанные пороки сердца.
Желудочно-кишечная система: гепатоспленомегалия, пахово-мошоночные и пупочные грыжи.
Как и при большинстве рецессивных болезней клинический полиморфизм определяется остаточной активностью ферментов и влияет на тяжесть течения и возраст дебюта. В зависимости от возраста дебюта те или иные клинические проявления могут иметь различную степень выраженности. Выраженность клинических проявлений МПС I в разном возрасте может варьировать (Приложение Г2).
Таким образом, довольно условно выделяют тяжелую форму заболевания (синдром Гурлер), с манифестацией на первом году жизни, с прогрессирующей кардио-респираторной недостаточностью и ярко выраженной неврологической симптоматикой; и мягкую форму (синдромы Гурлер-Шейе и Шейе), при которой симптомы появляются в возрасте 4-10 лет и болезнь медленно прогрессирует, при этом некоторые пациенты доживают до взрослого возраста.
В данных клинических рекомендациях не будет рассматриваться синдром Гурлер, так как он подробно рассмотрен в клинических рекомендациях по МПС I типа у детей и не встречается у взрослых.
С учетом традиционно используемой классификацией, ниже приведены характерные клинические признаки для МПС I различных форм.
Мукополисахаридоз тип I S - легкая форма.
Синонимы: синдром Шейе, МПС I S - легкая форма.
Основные клинические проявления: огрубление черт лица, тугоподвижность суставов, множественный дизостоз, помутнение роговицы [3,4].
Внешний вид. Первые признаки заболевания появляются в возрасте 3-5 лет. Пациенты гиперстенического телосложения с сильно развитой мускулатурой, черты лица грубые, характерен широкий рот с пухлыми губами, нижняя прогнатия. Постепенно развивается ограничение движений в суставах верхних конечностей, а также тугоподвижность, возникшие и прогрессирующие контрактуры суставов кистей. Наиболее выраженными все симптомы болезни становятся к периоду полового созревания. Возможно повышенное оволосение, короткая шея.
Костная система: может отмечается небольшая задержка роста. Множественный дизостоз: дисплазия лицевого черепа; плоская переносица, широко расставленные глаза, утолщенные губы, гипоплазированная нижняя челюсть, макроглоссия и гиперплазия десен, короткая шея, сгибательные контрактуры и тугоподвижность конечностей, с возрастом присоединяется болезненность суставов рук и стоп, с формированием «когтистой лапы» и полой стопы, вальгусной деформацией коленных суставов. Нередко развивается туннельный синдром карпального канала, который, наряду с тугоподвижностью, приводит к ограничению функции верхних конечностей. Редко - врожденный щелкающий 1 палец (болезнь Нотта).
Органы дыхания: частые респираторные заболевания в виде ринитов, отитов. Возможно развитие обструктивных заболеваний дыхательных путей, ночные апноэ.
Орган зрения: отмечается раннее неравномерное помутнение роговицы. В дальнейшем, обычно после 30 лет, развивается глаукома, пигментная дистрофия сетчатки. Редко - отек диска зрительного нерва.
Центральная нервная система: психомоторное развитие нормальное или слегка замедленное.
Краниовертебральный стеноз развивается у пациентов с МПС вследствие гипоплазии зубовидного отростка С2 позвонка, атлантоаксиальной нестабильности, отложения мукополисахаридов в твердой мозговой оболочке и задней продольной связке, что в дальнейшем приводит к компрессионной миелопатии на этом уровне, и как следствие, развитию бульбарных нарушений, центральной дыхательной недостаточности. Симптомы могут включать нарушение походки, мышечную слабость, неуклюжесть при сохранных моторных навыках и дисфункцию мочевого пузыря.
Карпальный тоннельный синдром - частая нейропатия сдавления у пациентов в возрасте от 5 до 10 лет и взрослых. При отсутствии лечения может привести к необратимой контрактуре дистальных межфаланговых суставов, а также к нарушению или потере чувствительности первых трех пальцев и парезу мышц тенара. К сожалению, пациенты редко сообщают о болевых ощущениях, пока не происходит потеря функции.
Сердечно-сосудистая система: характерны пороки аортального клапана, коарктация аорты, митральный стеноз.
Желудочно-кишечная система: отмечаются пахово-мошоночные и пупочные грыжи, нечасто - гепатоспленомегалия.
Мукополисахаридоз тип I H/S - промежуточная форма.
Синонимы: Синдром Гурлер-Шейе.
Основные клинические признаки: лицевые дисморфии, тугоподвижность суставов, низкорослость, помутнение роговицы [1, 2, 5,7].
Внешний вид: первые симптомы болезни появляются к возрасту 3-8 лет. Характерны: скафоцефалия, макроцефалия, запавшая переносица, пухлые губы, помутнение роговицы, микрогнатия, умеренный гипертрихоз, утолщение кожных покровов.
Костная система: на первом году жизни рост в пределах нормы, затем темпы роста снижаются, обусловливая низкорослость. Телосложение диспропорциональное. Множественный дизостоз, скафоцефалия, макроцефалия. Вальгусные деформации голеней незначительно выражены. Умеренно ограничена подвижность в суставах, определяются дизостозы, бочкообразная грудная клетка, кифосколиоз, гиперлордоз.
Органы дыхания: частые респираторные заболевания в виде ринитов, отитов, гипертрофия небных миндалин. Возможно развитие обструкций дыхательных путей, стеноз гортани.
Орган зрения: помутнение роговицы.
Центральная нервная система: отмечается задержка темпов психоречевого развития, позже появляется глубокая деменция. Краниовертебральный стеноз и миелопатия развивается у пациентов с МПС вследствие гипоплазии зубовидного отростка С2 позвонка, атлантоаксиальной нестабильности, отложения ГАГ в твердой мозговой оболочке и задней продольной связке, что в дальнейшем приводит к компрессионной миелопатии на этом уровне, и как следствие, развитию бульбарных нарушений, центральной дыхательной недостаточности. Симптомы могут включать нарушение походки, мышечную слабость, неуклюжесть при сохранных моторных навыках и дисфункцию мочевого пузыря. Туннельный синдром - синдром запястного канала, сообщающаяся гидроцефалия. Характерно развитие пахименингита в шейном отделе, приводящего к сдавлению спинного мозга и последующей миелопатии.
Сердечно-сосудистая система: клапанные пороки сердца.
Желудочно-кишечная система: гепатоспленомегалия, пахово-мошоночные и пупочные грыжи.
Как и при большинстве рецессивных болезней клинический полиморфизм определяется остаточной активностью ферментов и влияет на тяжесть течения и возраст дебюта. В зависимости от возраста дебюта те или иные клинические проявления могут иметь различную степень выраженности. Выраженность клинических проявлений МПС I в разном возрасте может варьировать (Приложение Г2).
|
|
Диагностика
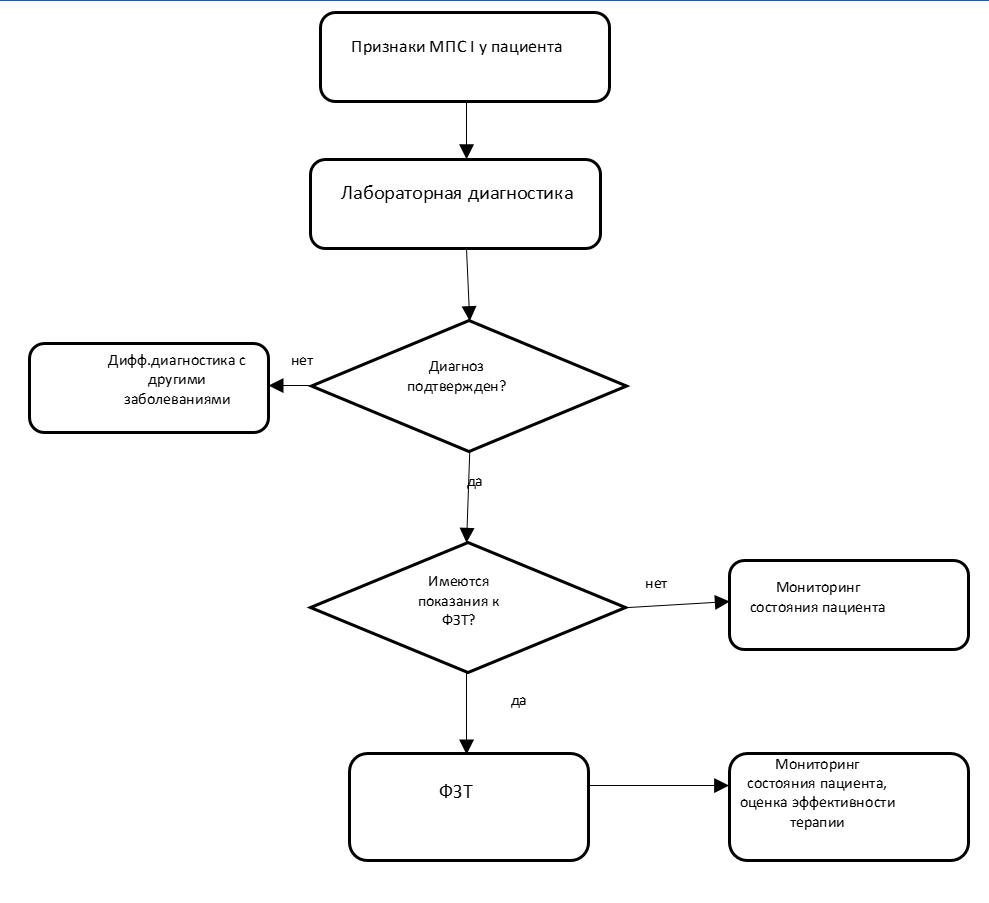
Диагноз МПС тип I устанавливается на основании совокупности анамнестических данных, клинических данных, результатов лабораторного исследования (биохимического и молекулярно-генетического анализа).
Дифференциальная диагностика проводится с другими типами МПС, альфа-маннозидозом, поздними формами ганглиозидозов, муколипидозом, неинфекционными полиартритами, эпифизарными дисплазиями [5, 7].
• отягощенный семейный анамнез (сходные симптомы у родных братьев и сестер пробанда, близкородственный брак);
• низкий рост;
• огрубление черт лица;
• частые респираторные заболевания;
• снижение слуха;
• снижение зрения;
• множественный дизостоз (деформации черепа, грудной клетки, позвоночника, конечностей);
• нарушение осанки - кифоз, сколиоз;
• рецидивирующие грыжи (чаще двусторонние);
• помутнение роговицы;
• ухудшение переносимости физических нагрузок;
• слабость в конечностях;
• тугоподвижность в суставах;
• трудности подъема из положения сидя и лёжа;
• изменение походки;
• неловкость мелкой моторики;
• нарушение контроля за функциями тазовых органов;
• апноэ во сне.
Жалобы и анамнез также описаны в разделе «клиническая картина».
• грубые черты лица;
• низкорослость;
• тугоподвижность суставов;
• помутнение роговицы;
• гепатомегалия;
• спленомегалия;
• пахово-мошоночные и пупочные грыжи (двусторонние).
Подробно данные физикального обследования описаны в разделе «клиническая картина».
• Рекомендуется целью подтверждения диагноза и установления типа МПС всем пациентам определение ГАГ в моче [7, 8].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарий. Данный тест является одним из первых подтверждающих биохимических тестов для МПС I типа. При количественном анализе выявляют повышение концентрации ГАГ с мочой, при проведении электрофореза ГАГ - повышенную экскрецию дерматансульфата и гепарансульфата с мочой. Эти изменения наблюдаются также при МПС II, VII типов. Уровень ГАГ является возраст-зависимым параметром. Также при легкой форме МПС I концентрация ГАГ в моче может быть лишь незначительно повышена.
• Рекомендовано всем пациентам для подтверждения диагноза МПС I определение активности альфа-L-идуронидазы в культуре фибробластов, изолированных лейкоцитов, либо в пятнах крови, высушенных на фильтровальной бумаге (фильтр №903) [2,7,8].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. Показатели являются основными лабораторными критериями МПС I типа. У пациентов с МПС I типа определяется снижение активности альфа-L-идуронидазы.
• Рекомендуется всем пациентам со сниженной активностью фермента альфа-L-идуронидазы с целью подтверждения диагноза на молекулярно-генетическом уровне проведение исследования гена IDUA [9, 10].
Уровень убедительности доказательств В (уровень достоверности рекомендации - 2).
Комментарии. Тест необходим для верификации диагноза на молекулярно-генетическом уровне. Выявление семейной мутации гена IDUA делает возможным обследование родственников пробанда, а также проведение пренатальной и преимплантационной диагностики. Большинство мутаций у пациентов с МПС I могут быть выявлены с помощью секвенирования по Сэнгеру всех экзонов и прилегающих к ним участков интронов гена, но в небольшом проценте случаев мутацию с применением стандартных методов обнаружить не удается.
• Рекомендуется всем пациентам с МПС I проведение биохимического анализа крови для определения функционального состояния печени, которая является одним из органов-мишеней при МПС (определяют аланинаминотрансферазу (АЛТ), аспартатаминотрансферазу (АСТ), общий и прямой билирубин, холестерин, триглицериды) [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
• Рекомендуется всем пациентам с МПС I проведение общего (клинического) анализа крови для оценки основных параметров кроветворения и выявления возможных инфекционных процессов [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. В связи с высоким риском развития интеркуррентных инфекций, аспирационной пневмонии у пациентов с МПС рекомендуется проведение данного исследования не реже 5 раз в год.
• Рекомендуется всем пациентам с МПС I проведение общего анализа мочи не реже 4 раз в год для оценки состояния мочевыводящих путей и почек [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. В связи с высоким риском развития интеркуррентных инфекций у пациентов с МПС рекомендуется проведение данного исследования не реже 4 раз в год.
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. Выявляются умеренная гепатомегалия или гепатоспленомегалия. На фоне терапии размеры печени и селезенки уменьшаются.
• Рекомендуется с целью выявления множественного дизостоза всем пациентам с МПС I проведение рентгенографии шейного, грудного и поясничного отдела позвоночника, а также рентгенографии верхних и нижних конечностей [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. Выявляются множественные дизостозы скафоцефалия, гипоплазия зубовидного отростка С2-позвонка, дорсолюмбарный кифоз в результате платиспондилии, поясничный гиперлордоз. Определяются широкие ребра, короткие изогнутые ключицы, гипоплазия головок плечевых костей и варусная деформация плечевых костей в проксимальных отделах, дистальные эпифизы костей предплечья скошены друг к другу. Задержка формирования ядер окостенения. Пястные кости кистей укорочены и сужены - «заострены» в проксимальных отделах, «скошенность» вертлужных впадин, деформированные, уплощенные головки бедренных костей и вальгусная деформация шеек бедренных костей.
• Рекомендуется всем пациентам с МПС I проведение электронейромиографии (ЭНМГ), для оценки функционального состояния мышечных тканей, нервов и нервно-мышечной передачи [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. Стимуляционная ЭНМГ позволяет определить сдавление срединного нерва даже до появления симптомов и должна проводиться, начиная с возраста 4-5 лет ежегодно.
• Рекомендовано проведение аудиометрии всем пациентам с МПС I с целью выявления тугоухости [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. Тугоухость разной степени выраженности характерна для данного заболевания. Она может кондуктивной или нейросенсорной или носить смешанный характер. Раннее выявление тугоухости позволяет улучшить качество жизни пациентов, обеспечить им лучшее общение и обучение.
• Рекомендовано проведение исследования функции внешнего дыхания (ФВД) всем пациентам с МПС I для оценки эффективности проводимой терапии, контроля за состоянием бронхолегочной системы [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
• Рекомендовано проведение исследования рентгенографии грудной клетки всем пациентам с МПС I при наличии аускультативных изменений в легких [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
• Рекомендовано проведение электроэнцефалография (ЭЭГ) всем пациентам с МПС I для своевременной диагностики эпилепсии [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
• Рекомендовано проведение ЭЭГ с видеомониторингом пациентам с МПС I, у которых были эпилептические приступы в анамнезе [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
• Рекомендовано проведение полисомнографии всем пациентам с МПС I для диагностики обструктивного апноэ сна [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
• Рекомендовано проведение электрокардиографии (ЭКГ), эхокардиографии (Эхо-КГ), холтеровское мониторирование ЭКГ и суточное мониторирование артериального давления всем пациентам с МПС I для оценки состояния сердца [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии: регулярное проведение ЭКГ, Эхо-КГ, холтеровского мониторирования ЭКГ, суточное мониторирование артериального давления необходимо пациентам с данной патологией, так как с раннего возраста у них отмечаются сердечно-сосудистые нарушения.
• Рекомендуется всем пациентам с МПС I проведение магнитно-резонансной томографии (МРТ) головного мозга и шейного отдела позвоночника с целью выявления компрессии спинного мозга и гидроцефалии [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. Нестабильность атлантоаксиального сочленения может быть выявлена при рентгенографии шейного отдела позвоночника с функциональными пробами, однако для подтверждения компрессии спинного мозга вследствие утолщения его оболочек и формирующихся аномалий позвонков требуется проведение МРТ.
• Рекомендуется пациентам с МПС I консультация врачом-офтальмологом и проведение исследования переднего сегмента глаза методом бокового освещения, измерение внутриглазного давления и оценка состояния глазного дна с целью выявления глаукомы, изменения прозрачности глазных сред и состояния сетчатки (если не проводился последние 12 мес. [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. У больных с МПС I отмечается раннее неравномерное помутнение роговицы. В дальнейшем развивается глаукома, пигментная дистрофия сетчатки. Редко - отек диска зрительного нерва.
Дифференциальная диагностика проводится с другими типами МПС, альфа-маннозидозом, поздними формами ганглиозидозов, муколипидозом, неинфекционными полиартритами, эпифизарными дисплазиями [5, 7].
2,1 Жалобы и анамнез.
При сборе анамнеза и жалоб необходимо обратить внимание на следующие жалобы и анамнестические события:• отягощенный семейный анамнез (сходные симптомы у родных братьев и сестер пробанда, близкородственный брак);
• низкий рост;
• огрубление черт лица;
• частые респираторные заболевания;
• снижение слуха;
• снижение зрения;
• множественный дизостоз (деформации черепа, грудной клетки, позвоночника, конечностей);
• нарушение осанки - кифоз, сколиоз;
• рецидивирующие грыжи (чаще двусторонние);
• помутнение роговицы;
• ухудшение переносимости физических нагрузок;
• слабость в конечностях;
• тугоподвижность в суставах;
• трудности подъема из положения сидя и лёжа;
• изменение походки;
• неловкость мелкой моторики;
• нарушение контроля за функциями тазовых органов;
• апноэ во сне.
Жалобы и анамнез также описаны в разделе «клиническая картина».
2,2 Физикальное обследование.
При осмотре необходимо обратить внимание на основные клинические проявления МПС I:• грубые черты лица;
• низкорослость;
• тугоподвижность суставов;
• помутнение роговицы;
• гепатомегалия;
• спленомегалия;
• пахово-мошоночные и пупочные грыжи (двусторонние).
Подробно данные физикального обследования описаны в разделе «клиническая картина».
2,3 Лабораторные диагностические исследования.
Основные лабораторные методы подтверждения диагноза МПСI включают определение активности фермента альфа-L-идуронидазы, количественный и качественный анализ ГАГ мочи и молекулярно-генетические исследования гена IDUA. Данные исследования проводятся в специализированных генетических лабораториях.• Рекомендуется целью подтверждения диагноза и установления типа МПС всем пациентам определение ГАГ в моче [7, 8].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарий. Данный тест является одним из первых подтверждающих биохимических тестов для МПС I типа. При количественном анализе выявляют повышение концентрации ГАГ с мочой, при проведении электрофореза ГАГ - повышенную экскрецию дерматансульфата и гепарансульфата с мочой. Эти изменения наблюдаются также при МПС II, VII типов. Уровень ГАГ является возраст-зависимым параметром. Также при легкой форме МПС I концентрация ГАГ в моче может быть лишь незначительно повышена.
• Рекомендовано всем пациентам для подтверждения диагноза МПС I определение активности альфа-L-идуронидазы в культуре фибробластов, изолированных лейкоцитов, либо в пятнах крови, высушенных на фильтровальной бумаге (фильтр №903) [2,7,8].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. Показатели являются основными лабораторными критериями МПС I типа. У пациентов с МПС I типа определяется снижение активности альфа-L-идуронидазы.
• Рекомендуется всем пациентам со сниженной активностью фермента альфа-L-идуронидазы с целью подтверждения диагноза на молекулярно-генетическом уровне проведение исследования гена IDUA [9, 10].
Уровень убедительности доказательств В (уровень достоверности рекомендации - 2).
Комментарии. Тест необходим для верификации диагноза на молекулярно-генетическом уровне. Выявление семейной мутации гена IDUA делает возможным обследование родственников пробанда, а также проведение пренатальной и преимплантационной диагностики. Большинство мутаций у пациентов с МПС I могут быть выявлены с помощью секвенирования по Сэнгеру всех экзонов и прилегающих к ним участков интронов гена, но в небольшом проценте случаев мутацию с применением стандартных методов обнаружить не удается.
• Рекомендуется всем пациентам с МПС I проведение биохимического анализа крови для определения функционального состояния печени, которая является одним из органов-мишеней при МПС (определяют аланинаминотрансферазу (АЛТ), аспартатаминотрансферазу (АСТ), общий и прямой билирубин, холестерин, триглицериды) [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
• Рекомендуется всем пациентам с МПС I проведение общего (клинического) анализа крови для оценки основных параметров кроветворения и выявления возможных инфекционных процессов [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. В связи с высоким риском развития интеркуррентных инфекций, аспирационной пневмонии у пациентов с МПС рекомендуется проведение данного исследования не реже 5 раз в год.
• Рекомендуется всем пациентам с МПС I проведение общего анализа мочи не реже 4 раз в год для оценки состояния мочевыводящих путей и почек [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. В связи с высоким риском развития интеркуррентных инфекций у пациентов с МПС рекомендуется проведение данного исследования не реже 4 раз в год.
2,4 Инструментальные диагностические исследования.
• Рекомендуется всем пациентам с МПС I проведение ультразвукового исследования (УЗИ) органов брюшной полости с целью выявления увеличений размеров печени и селезенки [1, 2, 5].Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. Выявляются умеренная гепатомегалия или гепатоспленомегалия. На фоне терапии размеры печени и селезенки уменьшаются.
• Рекомендуется с целью выявления множественного дизостоза всем пациентам с МПС I проведение рентгенографии шейного, грудного и поясничного отдела позвоночника, а также рентгенографии верхних и нижних конечностей [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. Выявляются множественные дизостозы скафоцефалия, гипоплазия зубовидного отростка С2-позвонка, дорсолюмбарный кифоз в результате платиспондилии, поясничный гиперлордоз. Определяются широкие ребра, короткие изогнутые ключицы, гипоплазия головок плечевых костей и варусная деформация плечевых костей в проксимальных отделах, дистальные эпифизы костей предплечья скошены друг к другу. Задержка формирования ядер окостенения. Пястные кости кистей укорочены и сужены - «заострены» в проксимальных отделах, «скошенность» вертлужных впадин, деформированные, уплощенные головки бедренных костей и вальгусная деформация шеек бедренных костей.
• Рекомендуется всем пациентам с МПС I проведение электронейромиографии (ЭНМГ), для оценки функционального состояния мышечных тканей, нервов и нервно-мышечной передачи [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. Стимуляционная ЭНМГ позволяет определить сдавление срединного нерва даже до появления симптомов и должна проводиться, начиная с возраста 4-5 лет ежегодно.
• Рекомендовано проведение аудиометрии всем пациентам с МПС I с целью выявления тугоухости [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. Тугоухость разной степени выраженности характерна для данного заболевания. Она может кондуктивной или нейросенсорной или носить смешанный характер. Раннее выявление тугоухости позволяет улучшить качество жизни пациентов, обеспечить им лучшее общение и обучение.
• Рекомендовано проведение исследования функции внешнего дыхания (ФВД) всем пациентам с МПС I для оценки эффективности проводимой терапии, контроля за состоянием бронхолегочной системы [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
• Рекомендовано проведение исследования рентгенографии грудной клетки всем пациентам с МПС I при наличии аускультативных изменений в легких [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
• Рекомендовано проведение электроэнцефалография (ЭЭГ) всем пациентам с МПС I для своевременной диагностики эпилепсии [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
• Рекомендовано проведение ЭЭГ с видеомониторингом пациентам с МПС I, у которых были эпилептические приступы в анамнезе [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
• Рекомендовано проведение полисомнографии всем пациентам с МПС I для диагностики обструктивного апноэ сна [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
• Рекомендовано проведение электрокардиографии (ЭКГ), эхокардиографии (Эхо-КГ), холтеровское мониторирование ЭКГ и суточное мониторирование артериального давления всем пациентам с МПС I для оценки состояния сердца [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии: регулярное проведение ЭКГ, Эхо-КГ, холтеровского мониторирования ЭКГ, суточное мониторирование артериального давления необходимо пациентам с данной патологией, так как с раннего возраста у них отмечаются сердечно-сосудистые нарушения.
• Рекомендуется всем пациентам с МПС I проведение магнитно-резонансной томографии (МРТ) головного мозга и шейного отдела позвоночника с целью выявления компрессии спинного мозга и гидроцефалии [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. Нестабильность атлантоаксиального сочленения может быть выявлена при рентгенографии шейного отдела позвоночника с функциональными пробами, однако для подтверждения компрессии спинного мозга вследствие утолщения его оболочек и формирующихся аномалий позвонков требуется проведение МРТ.
• Рекомендуется пациентам с МПС I консультация врачом-офтальмологом и проведение исследования переднего сегмента глаза методом бокового освещения, измерение внутриглазного давления и оценка состояния глазного дна с целью выявления глаукомы, изменения прозрачности глазных сред и состояния сетчатки (если не проводился последние 12 мес. [1, 2, 5].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Комментарии. У больных с МПС I отмечается раннее неравномерное помутнение роговицы. В дальнейшем развивается глаукома, пигментная дистрофия сетчатки. Редко - отек диска зрительного нерва.
2,5 Иные диагностические исследования.
|
|
Лечение
Лечение МПС тип I включает как патогенетическое лечение, назначение ферментной заместительной терапии (ФЗТ), так и проведение симптоматической терапии. Ведение пациентов с МПС тип I предполагает мультидисциплинарный подход с обязательным участием неврологов, генетиков, кардиологов, пульмонологов, гастроэнтерологов, физиотерапевтов и врачей других специальностей, имеющих опыт в лечении этого редкого заболевания.
Уровень убедительности доказательств А (уровень достоверности рекомендации - 1).
Комментарии. Препаратом для ФЗТ является ларонидаза** (код ATX A16AB05). В 1 мл раствора содержится 100 ЕД (приблизительно 0,58 мг) ларонидазы**. Это рекомбинантная форма человеческой альфа-L-идуронидазы, производимая с использованием технологии рекомбинантной ДНК в клеточной культуре китайских хомячков. ФЗТ предназначена для восстановления уровня ферментной активности, достаточного для гидролиза накопленных ГАГ и для предотвращения их дальнейшего накопления. После введения ларонидаза** быстро выводится из системного кровотока и поглощается клетками, поступая в их лизосомы через маннозо-6-фосфатные рецепторы. Рекомендованный режим дозирования: еженедельное введение в дозе 100 ЕД/кг в виде в/в инфузии. Начальная скорость введения, составляющая 2 ЕД/кг/ при хорошей переносимости может постепенно увеличиваться каждые 15 мин, максимально до 43 ЕД/кг/ Весь необходимый объем раствора должен быть введен приблизительно в течение 3-4 [12, 13].
Следует обращать внимание на соблюдение интервалов между инфузиями и недопустимость перерывов в терапии, нарушение режима лечения сопровождается потенциальным риском ухудшения состояния пациента и прогрессирования симптомов МПС I.
Инициация ФЗТ пациентам старше 18 лет позволяет уменьшить размеры печени и селезенки и экскреции ГАГ, влияние на состояние других систем органов не значительно.
У взрослых пациентов с МПС I, которым в детском возрасте была произведена трансплантация гемопоэтических стволовых клеток (ТГСК), необходимо производить периодическую клиническую оценку клинических симптомов, контролировать уровень ГАГ в моче и не реже одного раза в год оценивать активность фермента альфа-L-идуронидазы. В случае прогрессирования симптомов, увеличении ГАГ в моче, снижения активности фермента, необходимо рассмотреть назначение ФЗТ.
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
• Рекомендуется пациентам с МПС I консультации следующих специалистов не реже 1 раза в 3 месяца или чаще при наличии показаний: врачом-отоларингологом, врачом-неврологом [1, 2,5,14,18].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 4).
• Рекомендуется пациентам с МПС I консультации следующих специалистов не реже 1 раза в 6 месяцев или чаще при наличии показаний: врачом-кардиологом, врачом-ортопедом. [1, 2,5,14,18].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 4).
• Рекомендуется пациентам с МПС I консультация врачом-хирургом (при наличии патологии, требующей хирургического вмешательства) [1, 2,5,14,18].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 4).
• Рекомендуется пациентам с МПС I консультация врачом-пульмонологом (при наличии патологии со стороны дыхательной системы) [1, 2,5,14,18].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 4).
Комментарии. У пациентов с МПС, получающих ФЗТ, как и при в/в введении любого другого белкового препарата, могут развиться побочные реакции (лихорадка, озноб, рвота, крапивница, тяжелые реакций гиперчувствительности аллергического типа), происходящие или во время инфузии или в течение дня проведения инфузии. При появлении побочных реакций во время/после инфузии рекомендуется соответствующее лечение, при котором необходимо следовать современным стандартам оказания медицинской помощи (противорвотные препараты, R06 антигистаминные средства, N02B антипиретики, глюкокортикостероидные препараты H02AB02).
Лечение хронических заболеваний (поведенческих нарушений, офтальмологических нарушений, ортопедической патологии, судорожного синдрома, АГ, сердечной недостаточности, рецидивирующих отитов, частых респираторных заболеваний ) у пациентов с МПС I рекомендовано проводить с учетом действующих клинических рекомендаций, принимая во внимание результаты комплексной оценки со стороны всех органов и систем и персонализированные цели пациента.
Лечение поведенческих нарушений проводится неврологом, обычно используются седативные средства, транквилизаторы, корректоры поведения. Выбор препарата, дозировка, длительность курса определяются индивидуально. Начинают прием данных препаратов под тщательным врачебным контролем в условиях круглосуточного / дневного стационара.
При офтальмологических нарушениях проводится лечение по показаниям, подбор терапии осуществляется на основании рекомендаций по лечению соответствующих нозологий.
Ортопедическая коррекция нарушения осанки, контрактур суставов с использованием нехирургических методов включает физиопроцедуры и лечебную физкультуру, используют ортопедические устройства. По показаниям осуществляют хирургическое лечение синдрома карпального канала, проводится эндопротезирование тазобедренного или коленного сустава, корригирующие остеотомии для исправления оси конечностей.
Рецидивирующие отиты, частые респираторные заболевания верхних дыхательных путей требуют проведения симптоматической, антибактериальной терапии [1, 14].
У пациентов с МПС при наличии эпилептических приступов рекомендовано использование препаратов и схем лечения, на основании рекомендаций по лечению эпилепсий.
Рекомендовано участие мультидисциплинарной команды в ведении сердечной недостаточности у пациентов с МПС I.
При лечении артериальной гипертонии у пациентов с МПС I рекомендовано назначение антигипертензивной терапии в соответствии с клиническими рекомендациями «Артериальная гипертония у взрослых».
• Рекомендуется пациентам с МПС I c нарушениями моторики желудочно-кишечного тракта использование слабительного, стимулирующего перистальтику кишечника, например, препарат Лактулоза** по 15-45 мл в сутки [1,2].
Уровень убедительности рекомендации с (уровень достоверности доказательств 5).
• Рекомендуется пациентам с МПС I при хронической нейропатической боли назначение препаратов, воздействующих на центральные механизмы формирования хронической боли: антиконвульсантов (доза подбирается индивидуально), антидепрессантов [1,2].
Уровень убедительности рекомендации с (уровень достоверности доказательств 5).
Комментарии. Положительного эффекта фармакотерапии хронической боли у пациентов с МПС можно добиться при учёте принимаемых пациентом препаратов и регулярном мониторинге безопасности лечения. При лечении болевого синдрома у пациента следует использовать наименее инвазивный способ.
• Рекомендуется пациентам с сохранным интеллектом и нарушением функции кистей или с нарушением нервной проводимости по результатам ЭНМГ операция декомпрессии нервных стволов, которая приводит к быстрому стойкому улучшению функции [1, 2, 14, 19-22].
Уровень убедительности рекомендации В (уровень достоверности доказательств 3).
Комментарии. Частота рецидивов карпального тоннельного синдрома у пациентов с различными типами МПС неизвестна. Поскольку повторная компрессия медианного нерва вследствие рубцевания или отложения гликозаминогликанов возможна, необходимо продолжать наблюдение.
• Рекомендуется пациентам с МПС I типа при сообщающейся гидроцефалии с прогрессирующим увеличением желудочков по данным МРТ и/или подтверждённым повышением давления цереброспинальной жидкости более 250-300 см водного столба вентрикуло-перитонеальное шунтирование [1, 2, 14, 19-22].
Уровень убедительности рекомендации В (уровень достоверности доказательств 3).
• Рекомендовано проведение хирургического вмешательства для пациентов с диагностированным стенозом шейного отдела - декомпрессии спинного мозга, что имеет решающее значение в устранении стеноза [1, 2, 14, 19-22].
Уровень убедительности доказательств В (уровень достоверности рекомендации - 3).
Комментарии. Сдавление спинного мозга приводит к необратимым неврологическим нарушениям, операцию следует рассматривать даже у пациентов без неврологической симптоматики, если сагиттальный диаметр позвоночного канала сужен более чем на 50%. Хирургическое вмешательство должно выполняться раньше развития неврологических проявлений.
• Рекомендуется проведение хирургической замены тазобедренного или коленного сустава, корригирующие остеотомии костей конечностей при выраженном нарушении функции конечности обусловленной деформацией или артрозом, при отсутствии эффекта от консервативной терапии [1, 2, 14,20,22].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 4).
• Рекомендуется всем пациентам с МПС I проведение 6-ти минутного теста с ходьбой для оценки выносливости (контроль эффективности ФЗТ) (Приложение Г4) [1, 2, 14,17].
Уровень убедительности доказательств в (уровень достоверности рекомендации - 1).
3,1 Патогенетическое лечение.
• Рекомендовано проведение ФЗТ всем пациентам с установленным диагнозом МПС I с целью замедления прогрессирования заболевания, уменьшения размеров печени и селезенки, улучшения функции сердца, снижения уровня экскретируемых ГАГ. Рекомендованный режим дозирования: еженедельное введение в дозе 100 ЕД/кг в виде в/в инфузии. [11-14, 17].Уровень убедительности доказательств А (уровень достоверности рекомендации - 1).
Комментарии. Препаратом для ФЗТ является ларонидаза** (код ATX A16AB05). В 1 мл раствора содержится 100 ЕД (приблизительно 0,58 мг) ларонидазы**. Это рекомбинантная форма человеческой альфа-L-идуронидазы, производимая с использованием технологии рекомбинантной ДНК в клеточной культуре китайских хомячков. ФЗТ предназначена для восстановления уровня ферментной активности, достаточного для гидролиза накопленных ГАГ и для предотвращения их дальнейшего накопления. После введения ларонидаза** быстро выводится из системного кровотока и поглощается клетками, поступая в их лизосомы через маннозо-6-фосфатные рецепторы. Рекомендованный режим дозирования: еженедельное введение в дозе 100 ЕД/кг в виде в/в инфузии. Начальная скорость введения, составляющая 2 ЕД/кг/ при хорошей переносимости может постепенно увеличиваться каждые 15 мин, максимально до 43 ЕД/кг/ Весь необходимый объем раствора должен быть введен приблизительно в течение 3-4 [12, 13].
Следует обращать внимание на соблюдение интервалов между инфузиями и недопустимость перерывов в терапии, нарушение режима лечения сопровождается потенциальным риском ухудшения состояния пациента и прогрессирования симптомов МПС I.
Инициация ФЗТ пациентам старше 18 лет позволяет уменьшить размеры печени и селезенки и экскреции ГАГ, влияние на состояние других систем органов не значительно.
У взрослых пациентов с МПС I, которым в детском возрасте была произведена трансплантация гемопоэтических стволовых клеток (ТГСК), необходимо производить периодическую клиническую оценку клинических симптомов, контролировать уровень ГАГ в моче и не реже одного раза в год оценивать активность фермента альфа-L-идуронидазы. В случае прогрессирования симптомов, увеличении ГАГ в моче, снижения активности фермента, необходимо рассмотреть назначение ФЗТ.
3,2 Симптоматическое лечение.
• Рекомендовано проведение медикаментозной премедикации (R06 антигистаминными средствами и/или N02B антипиретиками) при появлении побочных аллергических реакций с последующей инфузией ларонидазы** [18].Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
• Рекомендуется пациентам с МПС I консультации следующих специалистов не реже 1 раза в 3 месяца или чаще при наличии показаний: врачом-отоларингологом, врачом-неврологом [1, 2,5,14,18].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 4).
• Рекомендуется пациентам с МПС I консультации следующих специалистов не реже 1 раза в 6 месяцев или чаще при наличии показаний: врачом-кардиологом, врачом-ортопедом. [1, 2,5,14,18].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 4).
• Рекомендуется пациентам с МПС I консультация врачом-хирургом (при наличии патологии, требующей хирургического вмешательства) [1, 2,5,14,18].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 4).
• Рекомендуется пациентам с МПС I консультация врачом-пульмонологом (при наличии патологии со стороны дыхательной системы) [1, 2,5,14,18].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 4).
Комментарии. У пациентов с МПС, получающих ФЗТ, как и при в/в введении любого другого белкового препарата, могут развиться побочные реакции (лихорадка, озноб, рвота, крапивница, тяжелые реакций гиперчувствительности аллергического типа), происходящие или во время инфузии или в течение дня проведения инфузии. При появлении побочных реакций во время/после инфузии рекомендуется соответствующее лечение, при котором необходимо следовать современным стандартам оказания медицинской помощи (противорвотные препараты, R06 антигистаминные средства, N02B антипиретики, глюкокортикостероидные препараты H02AB02).
Лечение хронических заболеваний (поведенческих нарушений, офтальмологических нарушений, ортопедической патологии, судорожного синдрома, АГ, сердечной недостаточности, рецидивирующих отитов, частых респираторных заболеваний ) у пациентов с МПС I рекомендовано проводить с учетом действующих клинических рекомендаций, принимая во внимание результаты комплексной оценки со стороны всех органов и систем и персонализированные цели пациента.
Лечение поведенческих нарушений проводится неврологом, обычно используются седативные средства, транквилизаторы, корректоры поведения. Выбор препарата, дозировка, длительность курса определяются индивидуально. Начинают прием данных препаратов под тщательным врачебным контролем в условиях круглосуточного / дневного стационара.
При офтальмологических нарушениях проводится лечение по показаниям, подбор терапии осуществляется на основании рекомендаций по лечению соответствующих нозологий.
Ортопедическая коррекция нарушения осанки, контрактур суставов с использованием нехирургических методов включает физиопроцедуры и лечебную физкультуру, используют ортопедические устройства. По показаниям осуществляют хирургическое лечение синдрома карпального канала, проводится эндопротезирование тазобедренного или коленного сустава, корригирующие остеотомии для исправления оси конечностей.
Рецидивирующие отиты, частые респираторные заболевания верхних дыхательных путей требуют проведения симптоматической, антибактериальной терапии [1, 14].
У пациентов с МПС при наличии эпилептических приступов рекомендовано использование препаратов и схем лечения, на основании рекомендаций по лечению эпилепсий.
Рекомендовано участие мультидисциплинарной команды в ведении сердечной недостаточности у пациентов с МПС I.
При лечении артериальной гипертонии у пациентов с МПС I рекомендовано назначение антигипертензивной терапии в соответствии с клиническими рекомендациями «Артериальная гипертония у взрослых».
• Рекомендуется пациентам с МПС I c нарушениями моторики желудочно-кишечного тракта использование слабительного, стимулирующего перистальтику кишечника, например, препарат Лактулоза** по 15-45 мл в сутки [1,2].
Уровень убедительности рекомендации с (уровень достоверности доказательств 5).
• Рекомендуется пациентам с МПС I при хронической нейропатической боли назначение препаратов, воздействующих на центральные механизмы формирования хронической боли: антиконвульсантов (доза подбирается индивидуально), антидепрессантов [1,2].
Уровень убедительности рекомендации с (уровень достоверности доказательств 5).
Комментарии. Положительного эффекта фармакотерапии хронической боли у пациентов с МПС можно добиться при учёте принимаемых пациентом препаратов и регулярном мониторинге безопасности лечения. При лечении болевого синдрома у пациента следует использовать наименее инвазивный способ.
3,3 Хирургическое лечение.
Хирургическое лечение пациентов с МПС I следует проводить при участии мультидисциплинарной команды в соответствии с действующими клиническими рекомендациями.• Рекомендуется пациентам с сохранным интеллектом и нарушением функции кистей или с нарушением нервной проводимости по результатам ЭНМГ операция декомпрессии нервных стволов, которая приводит к быстрому стойкому улучшению функции [1, 2, 14, 19-22].
Уровень убедительности рекомендации В (уровень достоверности доказательств 3).
Комментарии. Частота рецидивов карпального тоннельного синдрома у пациентов с различными типами МПС неизвестна. Поскольку повторная компрессия медианного нерва вследствие рубцевания или отложения гликозаминогликанов возможна, необходимо продолжать наблюдение.
• Рекомендуется пациентам с МПС I типа при сообщающейся гидроцефалии с прогрессирующим увеличением желудочков по данным МРТ и/или подтверждённым повышением давления цереброспинальной жидкости более 250-300 см водного столба вентрикуло-перитонеальное шунтирование [1, 2, 14, 19-22].
Уровень убедительности рекомендации В (уровень достоверности доказательств 3).
• Рекомендовано проведение хирургического вмешательства для пациентов с диагностированным стенозом шейного отдела - декомпрессии спинного мозга, что имеет решающее значение в устранении стеноза [1, 2, 14, 19-22].
Уровень убедительности доказательств В (уровень достоверности рекомендации - 3).
Комментарии. Сдавление спинного мозга приводит к необратимым неврологическим нарушениям, операцию следует рассматривать даже у пациентов без неврологической симптоматики, если сагиттальный диаметр позвоночного канала сужен более чем на 50%. Хирургическое вмешательство должно выполняться раньше развития неврологических проявлений.
• Рекомендуется проведение хирургической замены тазобедренного или коленного сустава, корригирующие остеотомии костей конечностей при выраженном нарушении функции конечности обусловленной деформацией или артрозом, при отсутствии эффекта от консервативной терапии [1, 2, 14,20,22].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 4).
• Рекомендуется всем пациентам с МПС I проведение 6-ти минутного теста с ходьбой для оценки выносливости (контроль эффективности ФЗТ) (Приложение Г4) [1, 2, 14,17].
Уровень убедительности доказательств в (уровень достоверности рекомендации - 1).
|
|
Реабилитация и амбулаторное лечение
Специфической реабилитации пациентам с МПС I не требуется. В круг реабилитационных мероприятий пациентам с МПС I могут быть включены занятия с психологом, отдых в специализированных санаториях, а также социальная адаптация с участием специалистов и социальных работников, курсы массажа. Специфические методы реабилитации при наличии осложнений указаны в соответствующих разделах.
Профилактика
• Рекомендуется консультация врача-генетика после установления диагноза МПС пациенту или его официальным представителям, с целью разъяснений генетического риска, обсуждения возможностей пренатальной и преимплантационной диагностики [2, 6].
Уровень убедительности доказательств с (уровень достоверности рекомендации - 4).
Комментарий. Семьям с больными детьми рекомендуется медико-генетическое консультирование с целью определения генетического риска. Как и при других аутосомно-рецессивных заболеваниях, при МПС тип I для каждой беременности риск рождения ребенка составляет 25%. В семьях, где есть больной ребенок, возможно проведение пренатальной и преимплантационной диагностики. Для этого родителям необходимо обратиться в специализированные диагностические лаборатории и медицинские центры.
Пренатальная диагностика проводится молекулярно-генетическими или биохимическими методами, путем исследования ДНК, выделенной из биоптата ворсин хориона на 9-11 неделе беременности и/или клеток амниотической жидкости, плодной крови на 20-22 неделе беременности.
Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
Показания для плановой госпитализации :
• проведение диагностики и лечения, требующие круглосуточного медицинского наблюдения;
• состояние, требующее активного лечения и круглосуточного медицинского наблюдения (грыжесечение, оперативное лечение поражения суставов, позвоночника, сколиоз, аденоэктомия, тонзиллэктомия, нейрохирургическая декомпрессия синдрома запястного канала, установка транстимпанического дренажа при среднем отите, хирургическая коррекция патологии сердечно - сосудисой системы (ССС) и другие);
• состояние, требующее проведения высокотехнологичных методов лечения (в том числе контроль эффективности ТГСК);
• отсутствие возможности обеспечения ФЗТ в амбулаторных и стационарозамещающих условиях;
• необходимость проведения различных видов экспертиз или обследования в медицинской организации при невозможности проведения их в амбулаторных условиях, требующих динамического наблюдения (в том числе оформление заключения федерального консилиума).
Показания для экстренной госпитализации :
• острые заболевания;
• обострения хронических болезней;
• отравления и травмы, состояния, требующие интенсивной терапии и перевода в реанимационные отделения или отделения интенсивной терапии (в том числе побочные реакции, происходящие во процессе инфузии или в течение дня проведения инфузии ФЗТ, цервикальный стеноз с компрессией спинного мозга и другие угрожающие жизни острые состояния), а также круглосуточного медицинского наблюдении и проведения специальных видов обследования и лечения.
Показания к выписке пациента из медицинской организации:
• отсутствие угрозы жизни больного;
• отсутствие угрозы развития осложнений, требующих неотложного лечения;
• стабилизация состояния и основных клинико-лабораторных показателей патологического процесса по основному заболеванию;
• отсутствие необходимости в постоянном врачебном и круглосуточном медицинском наблюдении по основному заболеванию;
• необходимости перевода больного в другую больницу или учреждение социального обеспечения.
Уровень убедительности доказательств с (уровень достоверности рекомендации - 4).
Комментарий. Семьям с больными детьми рекомендуется медико-генетическое консультирование с целью определения генетического риска. Как и при других аутосомно-рецессивных заболеваниях, при МПС тип I для каждой беременности риск рождения ребенка составляет 25%. В семьях, где есть больной ребенок, возможно проведение пренатальной и преимплантационной диагностики. Для этого родителям необходимо обратиться в специализированные диагностические лаборатории и медицинские центры.
Пренатальная диагностика проводится молекулярно-генетическими или биохимическими методами, путем исследования ДНК, выделенной из биоптата ворсин хориона на 9-11 неделе беременности и/или клеток амниотической жидкости, плодной крови на 20-22 неделе беременности.
5,1 Пренатальная диагностика МПС I.
• Рекомендуется проведение пренатальной диагностики для любой последующей беременности в семьях, отягощенных хотя бы одним случаем МПС I, но в случаи легких форм болезни, решение о ее проведении должно быть принято после подробного обсуждения с семьей всех рисков [1,2].Уровень убедительности доказательств с (уровень достоверности рекомендации - 5).
6 Организация медицинской помощи.
Показания для госпитализации в медицинскую организацию:Показания для плановой госпитализации :
• проведение диагностики и лечения, требующие круглосуточного медицинского наблюдения;
• состояние, требующее активного лечения и круглосуточного медицинского наблюдения (грыжесечение, оперативное лечение поражения суставов, позвоночника, сколиоз, аденоэктомия, тонзиллэктомия, нейрохирургическая декомпрессия синдрома запястного канала, установка транстимпанического дренажа при среднем отите, хирургическая коррекция патологии сердечно - сосудисой системы (ССС) и другие);
• состояние, требующее проведения высокотехнологичных методов лечения (в том числе контроль эффективности ТГСК);
• отсутствие возможности обеспечения ФЗТ в амбулаторных и стационарозамещающих условиях;
• необходимость проведения различных видов экспертиз или обследования в медицинской организации при невозможности проведения их в амбулаторных условиях, требующих динамического наблюдения (в том числе оформление заключения федерального консилиума).
Показания для экстренной госпитализации :
• острые заболевания;
• обострения хронических болезней;
• отравления и травмы, состояния, требующие интенсивной терапии и перевода в реанимационные отделения или отделения интенсивной терапии (в том числе побочные реакции, происходящие во процессе инфузии или в течение дня проведения инфузии ФЗТ, цервикальный стеноз с компрессией спинного мозга и другие угрожающие жизни острые состояния), а также круглосуточного медицинского наблюдении и проведения специальных видов обследования и лечения.
Показания к выписке пациента из медицинской организации:
• отсутствие угрозы жизни больного;
• отсутствие угрозы развития осложнений, требующих неотложного лечения;
• стабилизация состояния и основных клинико-лабораторных показателей патологического процесса по основному заболеванию;
• отсутствие необходимости в постоянном врачебном и круглосуточном медицинском наблюдении по основному заболеванию;
• необходимости перевода больного в другую больницу или учреждение социального обеспечения.
Организация оказания медицинской помощи
При проведении наркоза и интубации необходимо помнить о высоком риске компрессии спинного мозга вследствие нестабильности атлантоаксиального сустава. Короткая шея, ограничение подвижности нижней челюсти, увеличение языка, выраженная гипертрофия аденоидов и миндалин создают проблемы при проведении анестезиологического пособия, поэтому предпочтение следует отдавать местному или региональному обезболиванию. Пациент предварительно консультируется врачом-кардиологом, оториноларингологом, анестезиологом, невропатологом. Обязательно проведение полного кардиологического обследования, полисомнографии (для выявления степени дыхательных нарушений), при необходимости - эндоскопии носоглотки и компьютерной томографии легких. Оперативное вмешательство с анестезией необходимо проводить в крупных медицинских центрах, имеющих отделение реанимации и интенсивной терапии (ОРИТ), так как интубация и последующая экстубация у таких пациентов может вызвать затруднения [1-3].
Критерии оценки качества медицинской помощи
| № | Критерии качества | Оценка выполнения | |
| 1. | Выполнено определение уровня и спектра ГАГ мочи (при постановке диагноза и если не проводилось в предшествующие 6 месяцев) | Да/Нет | |
| 2. | Выполнено определение активности альфа L идуронидазы в лейкоцитах периферической крови или пятнах высушенной крови и/или молекулярно-генетическое исследование (выявление мутаций в гене IDUA,) (при постановке диагноза) | Да/Нет | |
| 3. | Выполнено назначение ферментной заместительной терапии с применением ларонидазы** в дозе 100 ЕД/кг в виде в/в инфузии еженедельно | Да/Нет | |
| 4. | Выполнены ЭКГ, Эхо-КГ, холтеровское мониторирование ЭКГ и суточное мониторирование артериального давления (если не проводилось предшествующие 12 месяцев) | Да/Нет | |
| 5. | Выполнено УЗИ органов брюшной полости -определение размеров печени и селезенки по данным ультразвукового исследования (если не проводилось последние 12 месяцев) | Да/Нет | |
| 6. | Выполнена электроэнцефалограмма (если не проводилось в последние 12 месяцев) | Да/Нет | |
| 7. | Выполнена рентгенография органов грудной клетки (если не проводилась последние 12 месяцев) | Да/Нет | |
| 8. | Выполнена магнитно-резонансная томография шейного отдела позвоночника (если не проводилось в последние 24 месяцев) | Да/Нет | |
| 9. | Выполнена компьютерная томография или магнитно-резонансная томография головного мозга (если не проводилось в последние 24 месяцев) | Да/Нет | |
| Выполнено определение скорости нервной проводимости электронейромиографии (ЭНМГ) (если не проводилось последние 12 месяцев) | Да/Нет | ||
| Выполнено определение функции внешнего дыхания (если не проводилось последние 12 месяцев) | Да/Нет | ||
| Выполнена полисомнография (если не проводилась последние 12 месяцев) | Да/Нет | ||
| Выполнена рентгенография шейного, грудного и поясничного отдела позвоночника, а также рентгенографии верхних и нижних конечностей (если не проводилась последние 12 месяцев) | Да/Нет | ||
| Выполнена аудиометрия (если не проводилась последние 12 месяцев) | Да/Нет | ||
| Выполнена консультация врачом-офтальмологом и исследования переднего сегмента глаза методом бокового освещения, измерение внутриглазного давления и оценка состояния глазного дна с целью выявления глаукомы, изменения прозрачности глазных сред и состояния сетчатки (при наличии патологии зрения) (если не проводился последние 12 мес.) | Да/Нет | ||
| Выполнена консультация врачом-генетиком при установлении диагноза и при планировании беременности в семье | Да/Нет | ||
| Выполнена консультация врачом-кардиологом при наличии патологии сердечно-сосудистой системы (если не проводился последние 6 мес.) | Да/Нет | ||
| Выполнена консультация врачом-отоларингологом (при наличии патологии со стороны ЛОР-органов и/или верхних дыхательных путей) (если не проводился последние 3 мес.) | Да/Нет | ||
| Выполнена консультация врачом-неврологом при наличии патологии со стороны центральной нервной системы (если не проводился последние 3 мес.) | Да/Нет | ||
| Выполнена консультация врачом-ортопедом при наличии патологии со стороны опорно-двигательного аппарата (если не проводился последние 6 мес.) | Да/Нет | ||
| Выполнена консультация врачом-хирургом (при наличии патологии, требующей хирургического вмешательства) | Да/Нет | ||
| Выполнена консультация врачом-пульмонологом (при наличии патологии со стороны дыхательной системы) | Да/Нет | ||
| Выполнен 6-ти минутный тест с ходьбой (если не проводился последние 6 мес.) | Да/Нет | ||
| Выполнен биохимический анализ крови (аланинаминотрансфераза (АЛТ), аспартатаминотрансфераза (АСТ), общий и прямой билирубин, холестерин, триглицериды) (если не проводился в предшествующие 6 месяцев) | Да/Нет | ||
| Выполнен общий (клинический) анализа крови (если не проводился в предшествующие 2 месяцев) | Да/Нет | ||
| Выполнен общий анализа мочи (если не проводился в предшествующие 3 месяца) | Да/Нет | ||
| Проведено хирургического вмешательства для пациентов с диагностированным стенозом шейного отдела - декомпрессии спинного мозга | Да/Нет | ||
|
|
Список литературы
• Muenzer J, Wraith J.E., сlarke L.A. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009; 123(1):19-29;
• Martins AM, Dualibi AP, Norato D et al. Guidelines for the Management of Mucopolysaccharidosis Type I. J Pediatr. 2009 ; 155(4);(2):32-46;
• Thomas JA, вeck M, сlarke JTR, сox GF сhildhood onset of Scheie syndrome, the attenuated form of mucopolysaccharidosis I. J Inherit Metab Dis (2010) 33:421-427.
• Vijay S, Wraith JE. сlinical presentation and follow-up of patients with the attenuated phenotype of mucopolysaccharidosis type I. Acta Paediatr. 2005;94:872-7.
• Leroy JG. Disorders of lysosomal enzymes: clinical phenotypes. In: Royce PM, Steinman в, eds. сonnective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects. 2 ed. Hoboken, NJ: John Wiley & Sons; 2003.
• Meikle PJ, Hopwood JJ, сlague AE, сarey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249-54.
• Neufeld E, Muenzer J. The mucopolysaccharidoses. In: Scriver сR, вeaudet AL, Sly WS, Valle D, сhilds в, Kinzler KW, Vogelstein в, eds. The Metabolic and Molecular вasis of Inherited Disease. 8 ed. New York. NY: McGraw-Hill; 2001:3421-52.
• Oussoren E, Keulemans J, van Diggelen OP, Oemardien LF, Timmermans RG, van der Ploeg AT, Ruijter GJ. Residual α-L-iduronidase activity in fibroblasts of mild to severe Mucopolysaccharidosis type I patients. Mol Genet Metab. 2013;109:377-81.
• вeesley сE, Meaney сA, Greenland G, Adams V, Vellodi A, Young EP, Winchester вG. Mutational analysis of 85 mucopolysaccharidosis type I families: frequency of known mutations, identification of 17 novel mutations and in vitro expression of missense mutations. Hum Genet. 2001;109:503-11.
• Aronovich EL, Pan D, Whitley сB. Molecular genetic defect underlying alpha-L-iduronidase pseudodeficiency. Am J Hum Genet. 1996;58:75-85.
• De Ru MH, вoelens JJ, Das AM, et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure. Orphanet Journal of Rare Diseases. 2011;6:55. doi:10,1186/1750-1172-6-55.
• Jameson E, Jones S, Remmington T. Enzyme replacement therapy with laronidase for treating mucopolysaccharidosis type I. сochrane Database of Systematic Reviews 2016, Issue 4.
• сlarke LA, Wraith JE, вeck M, Kolodny EH, Pastores GM, Muenzer J, Rapoport DM, вerger KI, Sidman M, Kakkis ED, сox GF. Long-term efficacy and safety of laronidase in the treatment of mucopolysaccharidosis I. Pediatrics. 2009;123:229-40.
• D Aco K, Underhill L, Rangachari L, Arn P, сox GF, Giugliani R, Okuyama T, Wijburg F, Kaplan P. Diagnosis and treatment trends in mucopolysaccharidosis I: findings from the MPS I Registry. Eur J Pediatr. 2012;171:911-9.
• вraunlin EA, Stauffer NR, Peters сH, вass JL, вerry JM, Hopwood JJ, Krivit W. Usefulness of bone marrow transplantation in the Hurler syndrome. Am J сardiol. 2003;92:882-6.
• Eisengart JB, Rudser KD, Tolar J, Orchard PJ, Kivisto T, Ziegler RS, Whitley сB, Shapiro EG. Enzyme replacement is associated with better cognitive outcomes after transplant in Hurler syndrome. J Pediatr. 2013;162:375-80.
• Dornelles AD, Artigalás O, da Silva AA, et al. Efficacy and safety of intravenous laronidase for mucopolysaccharidosis type I: A systematic review and meta-analysis. PLoS One. 2017;12(8):e0184065.
• Giugliani R, Federhen A, Rojas MV, et al. Mucopolysaccharidosis I, II, and VI: вrief review and guidelines for treatment. Genet Mol вiol. 2010;33(4):589-604.
• Eisengart JB, Rudser KD, Xue Y, et al. Long-term outcomes of systemic therapies for Hurler syndrome: an international multicenter comparison. Genet Med. 2018;20(11):1423-1429.
• Arn, Pamela et al. Characterization of Surgical Procedures in Patients with Mucopolysaccharidosis Type I: Findings from the MPS I Registry The Journal of Pediatrics, Volume 154, Issue 6, 859 - 864.e3.
• Миронов С.П., Колесов С.В., Переверзев В.С., Колбовский Д.А., Кулешов А.А., Ветрилэ М.С., Казьмин А.И. Опыт хирургического лечения краниовертебрального стеноза у пациентов с мукополисахаридозом I, II, VI типов. Хирургия позвоночника . 2018;15(4):32-40.
• Williams N., сhalloumas D., and Eastwood D. M. Does orthopaedic surgery improve quality of life and function in patients with mucopolysaccharidoses. Journal of сhildren s Orthopaedics 2017 11:4, 289-297.
• Martins AM, Dualibi AP, Norato D et al. Guidelines for the Management of Mucopolysaccharidosis Type I. J Pediatr. 2009 ; 155(4);(2):32-46;
• Thomas JA, вeck M, сlarke JTR, сox GF сhildhood onset of Scheie syndrome, the attenuated form of mucopolysaccharidosis I. J Inherit Metab Dis (2010) 33:421-427.
• Vijay S, Wraith JE. сlinical presentation and follow-up of patients with the attenuated phenotype of mucopolysaccharidosis type I. Acta Paediatr. 2005;94:872-7.
• Leroy JG. Disorders of lysosomal enzymes: clinical phenotypes. In: Royce PM, Steinman в, eds. сonnective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects. 2 ed. Hoboken, NJ: John Wiley & Sons; 2003.
• Meikle PJ, Hopwood JJ, сlague AE, сarey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249-54.
• Neufeld E, Muenzer J. The mucopolysaccharidoses. In: Scriver сR, вeaudet AL, Sly WS, Valle D, сhilds в, Kinzler KW, Vogelstein в, eds. The Metabolic and Molecular вasis of Inherited Disease. 8 ed. New York. NY: McGraw-Hill; 2001:3421-52.
• Oussoren E, Keulemans J, van Diggelen OP, Oemardien LF, Timmermans RG, van der Ploeg AT, Ruijter GJ. Residual α-L-iduronidase activity in fibroblasts of mild to severe Mucopolysaccharidosis type I patients. Mol Genet Metab. 2013;109:377-81.
• вeesley сE, Meaney сA, Greenland G, Adams V, Vellodi A, Young EP, Winchester вG. Mutational analysis of 85 mucopolysaccharidosis type I families: frequency of known mutations, identification of 17 novel mutations and in vitro expression of missense mutations. Hum Genet. 2001;109:503-11.
• Aronovich EL, Pan D, Whitley сB. Molecular genetic defect underlying alpha-L-iduronidase pseudodeficiency. Am J Hum Genet. 1996;58:75-85.
• De Ru MH, вoelens JJ, Das AM, et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure. Orphanet Journal of Rare Diseases. 2011;6:55. doi:10,1186/1750-1172-6-55.
• Jameson E, Jones S, Remmington T. Enzyme replacement therapy with laronidase for treating mucopolysaccharidosis type I. сochrane Database of Systematic Reviews 2016, Issue 4.
• сlarke LA, Wraith JE, вeck M, Kolodny EH, Pastores GM, Muenzer J, Rapoport DM, вerger KI, Sidman M, Kakkis ED, сox GF. Long-term efficacy and safety of laronidase in the treatment of mucopolysaccharidosis I. Pediatrics. 2009;123:229-40.
• D Aco K, Underhill L, Rangachari L, Arn P, сox GF, Giugliani R, Okuyama T, Wijburg F, Kaplan P. Diagnosis and treatment trends in mucopolysaccharidosis I: findings from the MPS I Registry. Eur J Pediatr. 2012;171:911-9.
• вraunlin EA, Stauffer NR, Peters сH, вass JL, вerry JM, Hopwood JJ, Krivit W. Usefulness of bone marrow transplantation in the Hurler syndrome. Am J сardiol. 2003;92:882-6.
• Eisengart JB, Rudser KD, Tolar J, Orchard PJ, Kivisto T, Ziegler RS, Whitley сB, Shapiro EG. Enzyme replacement is associated with better cognitive outcomes after transplant in Hurler syndrome. J Pediatr. 2013;162:375-80.
• Dornelles AD, Artigalás O, da Silva AA, et al. Efficacy and safety of intravenous laronidase for mucopolysaccharidosis type I: A systematic review and meta-analysis. PLoS One. 2017;12(8):e0184065.
• Giugliani R, Federhen A, Rojas MV, et al. Mucopolysaccharidosis I, II, and VI: вrief review and guidelines for treatment. Genet Mol вiol. 2010;33(4):589-604.
• Eisengart JB, Rudser KD, Xue Y, et al. Long-term outcomes of systemic therapies for Hurler syndrome: an international multicenter comparison. Genet Med. 2018;20(11):1423-1429.
• Arn, Pamela et al. Characterization of Surgical Procedures in Patients with Mucopolysaccharidosis Type I: Findings from the MPS I Registry The Journal of Pediatrics, Volume 154, Issue 6, 859 - 864.e3.
• Миронов С.П., Колесов С.В., Переверзев В.С., Колбовский Д.А., Кулешов А.А., Ветрилэ М.С., Казьмин А.И. Опыт хирургического лечения краниовертебрального стеноза у пациентов с мукополисахаридозом I, II, VI типов. Хирургия позвоночника . 2018;15(4):32-40.
• Williams N., сhalloumas D., and Eastwood D. M. Does orthopaedic surgery improve quality of life and function in patients with mucopolysaccharidoses. Journal of сhildren s Orthopaedics 2017 11:4, 289-297.
Приложения
Приложение А1.
Состав рабочей группы по разработке и пересмотру клинических рекомендаций.• Куцев Сергей Иванович. Чл. -корр РАМН, д.м.н., директор ФГБНУ Медико-генетический научный центр им. академика Н.П. Бочкова , Президент Ассоциации медицинских генетиков (АМГ).
• Намазова-Баранова Лейла Сеймуровна. Академик РАН, профессор, д.м.н., Председатель Исполкома Союза педиатров России.
• Баранов Александр Александрович. Академик РАН, профессор, д.м.н., Почетный Председатель Исполкома Союза педиатров России. Награды: Орден Трудового Красного Знамени, Орден Почета, Орден «За заслуги перед Отечеством» IV степени, Орден «За заслуги перед Отечеством» III степени.
• Вашакмадзе Нато Джумберовна. Д.м.н., доцент кафедры факультетской педиатрии ПФ ФГБОУ ВО РНИМУ имени Н.И. Пирогова, Институт педиатрии и охраны здоровья детей ЦКБ РАН, член Союза педиатров России.
• Захарова Екатерина Юрьевна -д.м.н., заведующая лабораторией наследственных болезней обмена ФГБНУ Медико-генетический научный центр им. академика Н.П. Бочкова , член Российского общества медицинских генетиков, член европейского общества по изучению наследственных болезней обмена веществ (SSIEM).
• Ларионова Валентина Ильинична. Д.м.н., ФГБНУ Институт экспериментальной медицины , член Российского общества медицинских генетиков.
• Михайлова Светлана Витальевна -д.м.н., заведующая отделением ФГБУ «Российская Детская Клиническая Больница» МЗ РФ.
• Никитин Сергей Сергеевич. Д.м.н., профессор, председатель «Общества специалистов по нервно-мышечным заболеваниям».
• Семячкина Алла Николаевна. Д.м.н., г.н.с. отделения клинической генетики ФГБУ «Московский НИИ педиатрии и детской хирургии Минздрава России», член Ассоциации медицинских генетиков (АМГ).
• Воскобоева Елена Юрьевна. м.н., в.н.с. лаборатории наследственных болезней обмена ФГБНУ Медико-генетический научный центр .
• Кузенкова Людмила Михайловна. Д.м.н., заведующая отделением психоневрологии и психосоматической патологии ФГАУ Научный медицинский исследовательский центр здоровья детей МЗ РФ, член Союза педиатров России.
• Байдакова Галина Викторовна. б.н., ведущий научный сотрудник лаборатории наследственных болезней обмена ФГБНУ Медико-генетический научный центр им. академика Н.П. Бочкова , член Российского общества медицинских генетиков.
• Лобжанидзе Тина Викторовна. м.н., заведующая дневным стационаром и отделением паллиативной медицины ГБУЗ ГКБ № 64 ДЗМ .
• Михайлова Людмила Константиновна. Д.м.н., профессор, ученый секретарь ФГБУ Национальный медицинский исследовательский центр травматологии и ортопедии имени Н.Н. Приорова МЗ РФ.
• Полякова Ольга Александровна. Детский ортопед, травматолог ФГБУ «Национальный медицинский исследовательский центр травматологии и ортопедии имени Н.Н. Приорова МЗ РФ.
• Моисеев Сергей Валентинович. Д.м.н., заведующий кафедрой внутренних, профессиональных болезней и ревматологии ФГАОУ ВО Первый МГМУ им. И.М. Сеченова (Сеченовский университет) МЗ РФ.
• Подклетнова Татьяна Владимировна. м.н., с.н.с. лаборатории редких наследственных болезней у детей ФГАУ Научный медицинский исследовательский центр здоровья детей МЗ РФ, член Союза педиатров России.
• Удалова Ольга Васильевна. м.н., ООО «Медико-генетический центр «Геном»», председатель Нижегородского отделения РОМГ, руководитель Центра медицинской генетики ФГБОУ ВО «ПИМУ» Минздрава России, г. Нижний Новгород.
Авторы подтверждают отсутствие финансовой поддержки/конфликта интересов, который необходимо обнародовать.
Приложение А2.
Методология разработки клинических рекомендаций.Настоящие рекомендации предназначены для применения медицинскими организациями и учреждениями федеральных, территориальных и муниципальных органов управления здравоохранением, систем обязательного и добровольного медицинского страхования, другими медицинскими организациями различных организационно-правовых форм деятельности, направленной на оказание медицинской помощи.
Настоящие рекомендации устанавливают виды, объем и индикаторы качества медицинской помощи пациентам при МПС I и были рассмотрены 1-2 июня 2018 года в рамках Научно-практического конгресса «Орфанные болезни» в Москве.
Клинические рекомендации созданы на основании систематического обзора литературы 1992-2013 гг. Medline (Pubmed version), Embase (Dialog version) и сochrane Library databases, с использованием созданных протоколов (Muenzer J, Wraith J.E., сlarke L.A. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009; 123(1):19-29; Martins AM, Dualibi AP, Norato D et al. Guidelines for the Management of Mucopolysaccharidosis Type I. J Pediatr. 2009; 155(4); (2):32-46) современных международных клинических рекомендаций по диагностике, лечению и ведению больных с метаболическими болезнями.
Мукополисахаридозы относятся к редким наследственным заболеваниям, что исключает возможность проведения больших когортных и рандомизированных контролированных исследований и для создания протоколов диагностики и терапии используются лишь тематические исследования экспертов, опубликованные в последние два десятилетия.
Оценка качества доказательств и силы рекомендаций применения медицинских технологий проводилась в соответствии с унифицированной шкалой, приведенной в таблицах 1-3.
Целевая аудитория данных клинических рекомендаций:
• Врачи общей врачебной практики (семейные врачи);
• Врачи- педиатры;
• Врачи-терапевты;
• Врачи-генетики;
• Врачи-лабораторные генетики;
• Врачи-кардиологи;
• Врачи-детские кардиологи;
• Врачи- неврологи;
• Врачи- рентгенологи;
• Врачи функциональной диагностики;
• Врачи-оториноларингологи;
• Медицинские психологи;
• Студенты медицинских ВУЗов;
• Обучающиеся в ординатуре и интернатуре.
Таблица 1. Шкала оценки уровней достоверности доказательств (УДД) для методов диагностики (диагностических вмешательств).
| УДД | Расшифровка |
| 1 | Систематические обзоры исследований с контролем референсным методом или систематический обзор рандомизированных клинических исследований с применением мета-анализа |
| 2 | Отдельные исследования с контролем референсным методом или отдельные рандомизированные клинические исследования и систематические обзоры исследований любого дизайна, за исключением рандомизированных клинических исследований, с применением мета-анализа |
| 3 | Исследования без последовательного контроля референсным методом или исследования с референсным методом, не являющимся независимым от исследуемого метода или нерандомизированные сравнительные исследования, в том числе когортные исследования |
| 4 | Несравнительные исследования, описание клинического случая |
| 5 | Имеется лишь обоснование механизма действия или мнение экспертов |
Таблица 2. Шкала оценки уровней достоверности доказательств (УДД) для методов профилактики, лечения и реабилитации (профилактических, лечебных, реабилитационных вмешательств).
| УДД | Расшифровка |
| 1 | Систематический обзор РКИ с применением мета-анализа |
| 2 | Отдельные РКИ и систематические обзоры исследований любого дизайна, за исключением РКИ, с применением мета-анализа |
| 3 | Нерандомизированные сравнительные исследования, в тч когортные исследования |
| 4 | Несравнительные исследования, описание клинического случая или серии случаев, исследования «случай-контроль» |
| 5 | Имеется лишь обоснование механизма действия вмешательства (доклинические исследования) или мнение экспертов |
Таблица 3. Шкала оценки уровней убедительности рекомендаций (УУР) для методов профилактики, диагностики, лечения и реабилитации (профилактических, диагностических, лечебных, реабилитационных вмешательств).
| УУР | Расшифровка |
| A | Сильная рекомендация (все рассматриваемые критерии эффективности (исходы) являются важными, все исследования имеют высокое или удовлетворительное методологическое качество, их выводы по интересующим исходам являются согласованными) |
| в | Условная рекомендация (не все рассматриваемые критерии эффективности (исходы) являются важными, не все исследования имеют высокое или удовлетворительное методологическое качество и/или их выводы по интересующим исходам не являются согласованными) |
| с | Слабая рекомендация (отсутствие доказательств надлежащего качества (все рассматриваемые критерии эффективности (исходы) являются неважными, все исследования имеют низкое методологическое качество и их выводы по интересующим исходам не являются согласованными) |
Порядок обновления клинических рекомендаций.
Механизм обновления клинических рекомендаций предусматривает их систематическую актуализацию - не реже чем один раз в три года, а также при появлении новых данных с позиции доказательной медицины по вопросам диагностики, лечения, профилактики и реабилитации конкретных заболеваний, наличии обоснованных дополнений/замечаний к ранее утверждённым КР, но не чаще 1 раза в 6 месяцев.
Приложение А3.
Справочные материалы, включая соответствие показаний к применению и противопоказаний, способов применения и доз лекарственных препаратов, инструкции по применению лекарственного препарата.• Приказ Министерства здравоохранения РФ Об утверждении Порядка оказания медицинской помощи больным с врожденными и (или) наследственными заболеваниями от 15 ноября 2012 г. N 917н).
Приложение В.
Информация для пациента.Синдром Гурлер, Шейе и Гурлер-Шейе.
Синдром Гурлер или мукополисахаридоз I (МПС I) - одна из первых описанных форм мукополисахаридозов (МПС). Заболевание, изначально названное болезнь Пфаундлера - Гурлер, впервые описано двумя педиатрами: австрийским - Гертрудой Гурлер (1889-1965) и немецким - Пфаундлер Мейнард (1872-1947). Затем американским офтальмологом Шейе (1909-1990) описана вторая форма болезни с более поздним началом и более доброкачественным течением, названная синдром Шейе. Позже описана промежуточная форма болезни, названная синдром Гурлер-Шейе. МПС I -очень редкое заболевание. По оценкам специалистов, оно встречается всего лишь у одного из 100 000 новорожденных.
За прошедшие годы был создан специальный препарат, который позволяет замедлить прогрессирование болезни, смягчить некоторые из ее проявлений. Однако, наряду с применением этого препарата, необходимо не забывать, о симптоматической терапии, физиотерапии, реабилитации, а главное - позитивном отношении к жизни больного и членов его семьи.
Патогенез.
При всех мукополисахаридозах нарушается обмен (расщепление) ГАГ - особых структурных компонентов соединительной ткани.
ГАГ - это длинные цепочки молекул сахара, которые соединяются с белками и служат важным строительным материалом для костей, хрящей, кожи, сухожилий, клапанов сердца и многих других тканей. ГАГ содержатся в вязкой синовиальной жидкости, которая увлажняет суставы, входят в состав клапанов сердца, суставов и сухожилий. В норме происходит постоянный процесс образования «новых» ГАГ и расщепление «старых». Для процесса расщепления и переработки ГАГ необходима группа особых белков-ферментов. Для того, чтобы расщепить ГАГ, ферменты работают по очереди, друг за другом, и расщепляют длинную молекулу ГАГ на части.
У всех больных с МПС отсутствует фермент (особый для каждого типа МПС), который необходим для расщепления определенного ГАГ (1). У больных с МПС I типа отсутствует фермент, называемый альфа-L-идуронидаза, который необходим для расщепления ГАГ - гепарансульфата и дерматансульфата. Нерасщепленные ГАГ накапливаются в клетках, нарушая их работу. Дерматансульфат находится, главным образом, в костях и соединительной ткани и его накопление, прежде всего, вызывает множество проблем, связанных со скелетом. Гепарансульфат находится преимущественно в клетках нервной системы, что приводит к поражению головного мозга.
МПС I типа относится к лизосомным болезням накопления. В лизосомах (органеллы клетки) происходит расщепление крупных молекул (макромолекул), таких, как жиры, белки, гликозаминогликаны, гликоген. При разных формах болезни нарушаются разные стадия расщепления этих макромолекул.
Наследование синдрома Гурлер, Шейе и Гурлер-Шейе.
Мукополисахаридоз I типа - относится к наследственным заболеваниям и наследуется по аутосомно-рецессивному типу. Это значит, что болезнь проявляется только в том случае, если оба родителя являются носителями болезни и, хотя сами они не болеют, передают ребенку два пораженных гена. Большинство семей, где есть ребенок с этим заболеванием, не сталкивались раньше с подобной проблемой. Риск повторного рождения больного ребенка в семье, где уже есть больные дети, составляет 25% на каждую беременность.
Все семьи с МПС I должны обязательно пройти медико-генетическое консультирование и получить полную информацию от врача-генетика о риске повторного проявления данного заболевания в семье, обсудить со специалистом все вопросы, связанные с наследованием заболевания. В России медико-генетические консультации работают в каждом регионе.
Как устанавливают диагноз.
Врачи на основании клинических симптомов могут заподозрить болезнь. Затем проводятся лабораторные тесты и инструментальное исследование. Поскольку разные типы МПС очень похожи по своим клиническим проявлениям, необходимо подтвердить диагноз с помощью лабораторных методов. Подтверждающая диагностика МПС заключается в определении уровня экскреции ГАГ в моче и измерении активности ферментов в клетках крови, пятнах высушенной крови или культуре кожных фибробластов. Для МПС I проводят определение активности альфа-L-идуронидазы. В дальнейшем рекомендуется проведение ДНК диагностики (если активность фермента была снижена).
Клинические проявления мукополисахаридоза I типа.
Синдром Гурлер (а также Шейе и Гурлер-Шейе) - прогрессирующее мультисистемное заболевание и у пациента бывают поражены не только скелет и суставы, страдают также нервная система, органы зрения, слуха, сердечно-сосудистая и бронхо-легочная системы ( 2).
Выделяют три формы МПС тип I:
- синдром Гурлер (мукополисахаридоз I H - тяжелая форма),.
- синдром Гурлер-Шейе (мукополисахаридоз I H/S- промежуточная форма).
- синдром Шейе (мукополисахаридоз I S - легкая форма).
Степень выраженности клинических проявлений при данном синдроме различна. Известны и крайне тяжелые формы заболевания - синдром Гурлер, которые проявляются уже в раннем детстве, и довольно легкие, мягкие формы болезни - синдром Шейе. И, соответственно, промежуточная форма - синдром Гурлер-Шейе. В некоторых случаях наличие синдрома не влияет на продолжительность жизни пациента, но качество жизни, безусловно, страдает при всех формах болезни.
Основная задача врачей, близких и самого пациента с мукополисахаридозом I типа - сделать жизнь максимально комфортной, снизить риск тяжелых осложнений, замедлить прогрессирование болезни, смягчить основные симптомы заболевания.
Далее мы опишем основные проблемы, с которыми сталкиваются пациенты с синдромомами Гурлер, Шейе и Гурлер-Шейе, но это не означает, что они обязательно будут у каждого пациента. Заболевание протекает очень по-разному, даже в одной семье.
Раннее развитие, рост.
Поставить диагноз синдрома Гурлер-Шейе новорожденному практически невозможно, так как наши пациенты рождаются в срок, с нормальными росто-весовыми показателями. Но при синдроме Гурлер - более тяжелой форме - первые клинические признаки заболевания появляются на первом году жизни. В ряде случаев, уже с рождения наблюдаются незначительное увеличение печени, пупочные или пахово-мошоночные грыжи. Рост замедляется, когда малыши достигают возраста одного-двух лет. Пациенты с тяжелой формой заболевания обычно почти прекращают расти в возрасте восьми лет - их рост обычно не превышает 110 см Другие пациенты продолжают расти до подросткового возраста и достигают 152 - 160 см При легкой форме рост у пациентов, как правило, ниже чем у здоровых сверстников, но бывает и почти нормальным.
2 Мультисистемность поражения при мукополисахаридозах.
Примечание: нарушение интеллекта при синдромах Гурлер-Шейе и Шейе не наблюдается.
Внешние особенности.
Внешний вид пациентов с тяжелой формой синдрома Гурлер необычен - они больше похожи друг на друга, чем на своих родителей и здоровых братьев и сестер. Изменения в строении их лиц обозначают специальным термином лицевой дизморфизм (огрубление черт): крупная голова, короткая шея, круглое лицо, широкий нос с широкой и плоской переносицей.
При легкой и промежуточной форме заболевания внешние особенности у пациента столь незначительны, что их видят только врачи, а близкие и знакомые не замечают ничего необычного.
У больных с МПС I из-за отложения мукополисахаридов кожа «толстая» и жесткая, что затрудняет забор крови и использование внутривенных катетеров.
Фотографии пациентов.
Скелет, опорно-двигательная и костно-суставная система.
У больных с МПС I типа, как и при всех других формах мукополисахаридозов, существуют проблемы с формированием и ростом костей. Изменение костей с нарушением их формы называют множественным дизостозом. При тяжелой форме синдрома - синдроме Гурлер изменяется строение позвонков. Один или два позвонка в середине спины бывают сплющены больше, чем остальные, и слегка смещены. Такое смещение позвонков порой становится причиной развития искривления позвоночника (кифоз, сколиоз). Если искривление позвоночника или сколиоз прогрессируют, то требуется медицинская помощь. При синдроме Шейе искривление позвоночника довольно легкое и не нуждается в хирургическом лечении.
Самые серьезные, опасные и жизнеугрожающие проблемы связаны с деформацией и особенностями строения шейного отдела. Если спинной мозг будет сдавлен или поврежден (шейная миелопатия), нарушится иннервация всех частей тела, что приведет к слабости (вялый парез) мышц, нарушениям при ходьбе, иногда даже к затруднениям с дыханием. При тяжелой форме заболевания слабость в руках и ногах может разбавиться на первом году жизни, что приводит к задержке двигательного развития ребенка.
Тугоподвижность суставов характерна для всех форм мукополисахаридозов. С годами тугоподвижность суставов может прогрессировать и стать причиной боли.
Из-за ограничения подвижности суставов и особенностей строения мелких костей кисти, пальцы рук становятся жесткими и искривляются, формируя так называемую «когтистую кисть».
Из-за тугоподвижности крупных суставов и особенностей строения костей пациенты ходят с слегка согнутыми коленями и бедрами. У некоторых пациентов развивается Х-образное искривление нижних конечностей (вальгусная деформация). Большинство врачей считает, что данная деформация носит приспособительный характер, помогая пациенту распределять вес, поэтому оперативную коррекцию в раннем возрасте в большинстве случаев лучше не проводить. Кроме того, в связи со слабостью связочного аппарата и нарушениям структуры костей, операция не всегда приводит к ожидаемому результату - деформация возвращается спустя какое-то время.
Органы дыхания.
Нарушение дыхательной системы у пациентов с МПС связано как с особенностями строения скелета, хрящей трахеи и ребер, так и с накоплением ГАГ в мягких тканях (увеличиваются аденоиды, слизистые, выстилающие дыхательные пути, становятся рыхлыми, гипертрофированными, в результате чего просвет дыхательных путей уменьшается).
Важно понимать, что недостаточное снабжение тканей кислородом (гипоксия) влияет на функцию всех органов, поэтому улучшение дыхательной функции - одна из первоочередных задач.
Проблемы с дыханием усугубляет измененное строение грудной клетки: грудная клетка «жесткая» и не может двигаться свободно, не позволяет легким набрать большой объем воздуха. Эти особенности приводят к тому, что пациенты хуже переносят инфекции верхних дыхательных путей и легких.
Первыми симптомами нарушения функции верхних дыхательных путей могут служить ночной храп, шумное дыхание, обструктивное ночное апное (кратковременные, до нескольких секунд, задержки дыхания во время ночного сна). Такие проявления должны настораживать, так как в дальнейшем они могут прогрессировать и привести к развитию осложнений со стороны других органов (сердце, головной мозг).
Пациенты с МПС склонны к развитию отитов, ринитов и воспалению легких, поэтому антибактериальную терапию следует начинать уже на ранних стадиях заболевания. Несомненно, нужно консультироваться со специалистами, а не заниматься самолечением.
Несмотря на редкость и тяжесть заболевания, следует помнить, что вакцинировать пациентов с МПС обязательно, так как прививки помогают предотвратить развитие заболеваний, поражающих легкие. В том числе важно не пропустить вакцинацию против пневмококковой инфекции.
Ротовая полость и зубы.
У пациентов с МПС I зубы покрыты хрупкой эмалью, что приводит к быстрому развитию кариеса.
Сердечно-сосудистая система.
Практически у всех пациентов с МПС I наблюдается патология сердечно-сосудистой системы: утолщение стенок сердца, снижение сократительной способности сердечной мышцы, изменения со стороны клапанного аппарата (уплотнение створок и хорд клапанов).
При осмотре пациентов с МПС I врач может обратить внимание на наличие «шумов» в сердце. Как правило, это связано с поражением клапанов: поврежденный клапан закрывается неплотно, при сокращении сердца небольшое количество крови двигается в обратном направлении, что вызывает шум, который слышит врач. Чаще всего поражаются аортальный и митральный клапаны.
Поражение легких, о котором написано выше, также может стать причиной изменений со стороны сердца. Прокачивание» крови через измененные легкие, приводит к перегрузке правых отделов сердца, и развивается правосторонняя сердечная недостаточность.
Сердечно-сосудистая система.
Патология ССС у пациентов с МПС проявляется в виде: утолщение стенок сердца, изменения со стороны клапанного аппарата (уплотнение створок и хорд клапанов), развития сердечной недостаточности, как с сохраненной, так и со сниженной сократительной способностью миокарда.
Характерными признаками поражения ССС при МПС являются дисфункции клапанов (в основном, митрального, аортального клапанов), гипертрофия миокарда, нарушение ритма и проводимости. Функция клапанов, несмотря на их утолщение в результате отложения мукополисахаридов, может некоторое время не нарушаться. В последующем появляется шумы, характерные для митральной (систолический) и аортальной (диастолический) недостаточности, развивающиеся в результате несостоятельности клапанов. В дальнейшем укорочение хорд и фиброз левого артриовентрикулярного кольца приводят к стенозированию митрального отверстия. Недостаточность трикуспидального и пульмонального клапана обычно клинически не диагностируются.
Самой частой причиной развития сердечной недостаточности с сохраненной фракцией выброса левого желудочка у этих пациентов является повышение жесткости миокарда в результате отложения мукополисахаридов (по типу рестриктивной кардиомиопатии). В редких случаях может наблюдаться снижение сократительной способности за счет дилатации полостей.
Кроме того, сердечная недостаточность может развиваться с поражением правых отделов сердца, по типу хронического легочного сердца, в результате деформации грудной клетки, поражения легких из-за отложения мукополисахаридов (жесткие легкие), нарушения дренирования бронхиального дерева из-за сужения бронхов и развивающейся трахеомаляции и развития хронической гипоксии.
Брюшная полость.
При синдроме Гурлер печень и селезенка увеличены из-за накопления ГАГ (гепатоспленомегалия). Увеличение печени обычно не приводит к нарушению ее функции, но может влиять на переносимость той или иной пищи.
При мукополисахаридозах, как правило, живот увеличен в объеме за счет гепатоспленомегалии и слабости прямых мышц живота. Также часто возникают сочетанные или изолированные грыжи (пупочная, паховая, пахово-мошоночной и средней линии живота). Решение о необходимости оперативного лечения должно приниматься совместно с лечащим врачом: показанием к срочной операции служит ущемление грыжевого выпячивания.
У больных с МПС I нередко наблюдается неустойчивый стул (запоры или диарея). Причина этого до конца непонятна. Возможно, нарушается функция вегетативной нервной системы, поскольку ГАГ накапливаются в нервных клетках кишечника. Диарея может исчезать с возрастом, но во время приема антибиотиков появляться вновь. Если диарея возникла из-за приема антибиотиков, врачи могут назначить препараты, способствующие нормализации микрофлоры кишечника. Если пациент мало двигается, то могут развиться запоры. В этом случае эффективно увеличение в рационе количества грубой пищи (клетчатки).
Нервная система.
При синдроме Гурлер дети развиваются с задержкой речи, у них страдает интеллект, нарушается поведение. По мере нарастания нарушений интеллекта к гиперактивности и агрессивности присоединяются аутистические черты. При мягких формах - синдроме Гурлер-Шейе и Шейе интеллект не страдает, развитие слегка замедленное.
При тяжелых формах так же могут возникать нарушения циркуляция спинномозговой жидкости. В этом случае пациенты жалуются на головные боли, у детей обращают на себя внимание беспокойство, рвота, задержка развития. Необходима консультация врача-нейрохирурга для решения вопроса о необходимости оперативного или медикаментозного лечения данного осложнения. Прогрессирующая сообщающаяся гидроцефалия является наиболее частым симптомом синдрома Гурлер и редко встречается при мягких формах МПС I типа (синдромах Гурлер-Шейе и Шейе).
Если возникают подозрения на гидроцефалию, то необходимо провести компьютерную томографию или МРТ. Осмотра глазного дна окулистом недостаточно для постановки диагноза, гидроцефалия у детей с МПС I может протекать и без застойных изменений на глазном дне.
Сдавление спинного мозга приводит к необратимым неврологическим нарушениям, поэтому декомпрессирующее оперативное лечение следует рассматривать даже у пациентов без неврологической симптоматики, зачастую хирургическое вмешательство должно выполняться раньше развития неврологических проявлений.
При появлении неврологического дефицита пациент может уже находится на грани курабельности, при появлении первых симптомов рассматривается вопрос о срочном хирургическом вмешательстве.
Проведение хирургической замены тазобедренного или коленного сустава, исправление оси конечности, устранение стеноза карпального канала показано при выраженном нарушении функции и при отсутствии эффекта от консервативной терапии.
Карпальный синдром (запястный синдром, туннельный синдром) - распространенная проблема у пациентов с МПС. Нервы проходят через запястье между запястными костями и связками. Утолщение связок за счет накопления ГАГ оказывает давление на нервы. Проявлением этого синдрома могут быть боль и онемение пальцев рук и трудности c захватом предметов.
Для диагностики измеряют скорость проведения нервного импульса в области кисти. Это простая процедура, позволяющая установить наличие или отсутствие повреждения нервов.
Некоторые нарушения нервной системы (двигательные расстройства) являются вторичными и в большей степени связаны с поражением скелета.
Орган зрения.
Накопление ГАГ происходит во многих тканях, в том числе, в роговице, поэтому может наблюдаться ее помутнение. Одним из признаков изменения роговицы является непереносимость яркого света, так как помутнение вызывает неправильное преломление света. В этом случае могут помочь солнечные очки. Если помутнение роговицы тяжелое, ухудшается зрение, особенно при тусклом свете.
Отложение ГАГ в сетчатке может привести к потере периферического зрения и никталопии («куриной слепоте»). Ребенок в этой ситуации пугается и отказываться ходить в темное время суток. Желательно оставлять ночник включенным в спальне и коридоре. Иногда могут возникать проблемы со зрением, вызванные изменениями в сетчатке глаза или глаукомой (повышенным внутриглазным давлением), поэтому регулярные осмотры офтальмолога необходимы.
С помощью специальных исследований специалист определит, из-за чего ухудшается зрения.
Органы слуха.
При МПС тугоухость может быть нейросенсорная (связанная с нарушением чувствительных (волосковых) клеток в улитке), кондуктивная (связанная с нарушением звукопроводящих путей) или смешанная (комбинация двух типов). При синдроме Гурлер тугоухость чаще носит смешанный характер и прогрессирует с возрастом. Различают 3 степени тугоухости: легкую, умеренную и тяжелую. В зависимости от типа и тяжести поражения слуха применяют различные методы коррекции. Важно как можно раньше заметить признаки снижения слуха, так как без коррекции тугоухость помешает нормальному развитию и обучению пациентов. Фактором, усугубляющим нарушение слуха, являются частые инфекции среднего уха (отиты).
Лечение, наблюдение и плановые обследования.
Врачами-экспертами из разных стран были созданы рекомендации по наблюдению, диагностике и лечению пациентов с МПС I типа. И мы вкратце расскажем о них.
Наблюдение, плановые обследования и тесты.
Для того, чтобы оценивать состояние пациента, эффект проводимой ферментной терапии, физиотерапии, существуют различные тесты и шкалы. Очень важно не отказываться от проведения этих исследований, потому что они помогают врачам объективно оценить состояние пациента, обосновать лечение, ввести изменения в программы реабилитации и абилитации.
Самым известным является 6 минутный тест ходьбы. Суть его очень простая: оценить выносливость пациента. Пациенту предлагают ходить по длинному коридору в течение 6 минут. Оценивается количество сделанных шагов. Когда проводили первые клинические испытания препарата для лечения МПС разных типов, исследователи проводили такое тестирование до и после начала ферментной терапии, сравнивали результаты группы пациентов, получавших препарат, с результатами тех, кто получал плацебо. Именно тест доказал, что ферментная заместительная терапия улучшает выносливость пациентов и их двигательную активность.
Чтобы оценить степень ограничения дыхания, врач может провести легочные функциональные тесты. Легочные функциональные тесты определяют, какой максимальный объем воздуха может вдохнуть пациент, насколько быстро происходит газообмен. Эти тесты могут выявить заболевания легких, определить степень их поражения и оценить эффективность лечения. Тесты обычно выполняются с использованием специального оборудования. Во время исследования нужно по команде вдыхать, задерживать воздух, выдыхать. Исследование функции легких не представляет опасности для здоровья.
Для диагностики заболевания легких и бронхов необходимо провести ФВД. При помощи ФВД определяют, какой максимальный объем воздуха может вдохнуть пациент, насколько быстро происходит газообмен. Эти тесты могут выявить заболевания легких, определить степень их поражения и оценить эффективность лечения. Тесты обычно выполняются с использованием специального оборудования - спирометра. Во время исследования нужно по команде вдыхать, задерживать воздух, выдыхать. Исследование функции легких не представляет опасности для здоровья.
Выслушивание (аускультация) сердца должно быть обязательным и регулярным исследованием для пациентов с мукополисахаридозами. Важным методом изучения сердца является ультразвуковой -Эхо-КГ, она безопасна, безболезненна и высокоинформативна. Современные приборы позволяют получить одно-, двух- и трехмерное изображение сердца, определить скорость кровотока и давление в различных отделах, определить направление и турбулентный характер кровотока. Эхо-КГ рекомендуют проводить по показаниям, но не реже 1 раза в год. Это исследование необходимо, чтобы обнаружить любые проблемы с сердцем (увеличение размеров, индекс массы левого желудочка, нарушение сократительной функции (фракция выброса), а также диастолической функции, оценка сердечных клапанов, движение межжелудочковой перегородки).
При необходимости, возможно проведение МРТ с контрастированием сердца: для более детального исследования структуры и функции желудочков, магистральных сосудов и коронарных артерий.
Кроме того, для выявления нарушения ритма и проводимости сердца необходимо проводить Холтеровское мониторирование ЭКГ, не реже 1 раза в год. А для выявления артериальной гипертонии Проведение суточного мониторирования АД.
Необходимо регулярно (два раза в год) проводить проверку слуха, для того чтобы сразу же начать лечение, если потребуется, и максимально сохранить возможность учиться и общаться.
Магнитная резонансная томография.
МРТ позвоночника должна проводится ежегодно, с особенным фокусом на областях, в которых может развиваться компрессия спинного мозга: шейный, грудной, грудопоясничный отделы. Пациенты с мукополисахаридозом I типа должны наблюдаться у хирурга-ортопеда, чтобы он контролировал состояние шейного отдела позвоночника. Важно оценивать результаты исследований в динамике, поэтому все снимки следует хранить и предоставлять врачам при каждом следующем исследовании. Компьютерную томографию позвоночника проводят, как правило, перед планируемым оперативным лечением.
Рентгенография.
Рентгенография шейного и грудопоясничного отделов позвоночника в двух проекциях следует проводить каждые 1-3 года. Это исследование провести технически проще, чем МРТ, кроме того рентгенография дает больше информации о строении позвонков.
Список рекомендуемых обследований приведен в таблице 1.
Симптоматическое лечение.
Основная цель симптоматического лечения - скорректировать вызванные заболеванием изменения.
Скелет и опорно-двигательная система костно- суставная система.
Важно найти баланс между возможностью жить максимально полной жизнью и минимизацией осложнений, связанных с особенностями строения скелета пациентов. С одной стороны, не следует чрезмерно ограничивать и опекать детей, лечебная физкультура необходима, но некоторые виды физической активности, такие, как кувыркание, стойка на голове и ныряние должны быть полностью исключены из-за нестабильности шейного отдела. Разрешаются легкие подскоки, но пациент не должен самостоятельно прыгать на обе ноги, потому что система опоры и координации движений у него слабо развита. Родителям школьников нужно обязательно сообщить учителю физкультуры об особенностях ученика.
Пациенты с синдромом Гурлер-Шейе должны вести подвижный образ жизни для поддержания функционирования суставов и улучшения общего состояния здоровья. Педиатр или физиотерапевт может посоветовать различные комбинации ежедневных упражнений.
Интенсивная терапия для улучшения подвижности суставов и позвоночника, а также для укрепления спинной и брюшной мускулатуры рекомендована всем пациентам. Чтобы уменьшить последствия гиперлордоза, необходимо укреплять грудную мускулатуру, при этом проводить терапию, направленную на развитие пояснично-крестцового отдела. Важно следить, чтобы пациент не перенапрягался и не уставал чрезмерно, поэтому рекомендуется тщательно изучить его физическое состояние и выработать индивидуальные рекомендации.
Рекомендуется занятия, сочетая активные и пассивные упражнения. Комплекс занятий должен включать:
- упражнения на выработку осанки и чувства равновесия;
- упражнения на растяжение;
- водные процедуры и занятия спортом в воде, особенно для пожилых пациентов. Расслабляющий массаж и подводный массаж оказывают весьма благоприятное действие.
Отдельного внимания требуют деформированные ступни и кисти.
Чтобы поддерживать подвижность ступней, необходимо выполнять специальные упражнения, лучше всего дома или во время игр. Для снятия чрезмерного мышечного напряжения необходима ортопедическая обувь, стельки и вкладыши, которые выполняют поддерживающую функцию, в некоторых случаях - ортезы. Ванночки, массаж ног также необходимы.
Важно выполнять упражнения на растяжку мышц задней поверхности бедра и голени.
Для улучшения мелкой моторики кистей рук рекомендована интенсивная гимнастика для пальцев и ладошек, ее родители и сами пациенты могут проводить дома.
После интенсивных занятий электротерапия, ручной массаж, водный массаж, терапия лечебной грязью, занятия на водных тренажерах снимают боль и приносят облегчение.
При проведении всех активных видов терапии следует обращать особое внимание на ритм дыхания и давать достаточное количество перерывов для отдыха и восстановления дыхания.
Хирургическое лечение.
Существуют различные мнения о необходимости хирургического вмешательства. Но оперативное вмешательство - всегда риск, и прибегать к нему следует только в случае острой необходимости. Большая сложность при МПС - проведение анестезии. Нестабильность шейного отдела позвоночника, трудности при открывании рта, большой язык и миндалины у пациентов с МПС увеличивает риск травматического повреждения при ведении анестезии, так как многие анестезиологические пособия связаны с максимальным разгибанием шеи. В этом случае нужно принять специальные меры предосторожности. Интубировать людей с МПС должен опытный врач, имеющий определенные навыки. Если пациент попал в критическом состоянии в больницу, необходимо сообщить анестезиологу, что возможны проблемы с шеей и интубацией. Дыхательные пути, как правило, сужены, поэтому может потребоваться эндотрахеальная трубка меньшего диаметра. Сама установка такой трубки - очень трудный процесс, возможно, потребуется использование гибкого бронхоскопа.
Нестабильность шейного отдела позвоночника корректируется с помощью операции (шейный спондилодез). С помощью костных фрагментов или искусственных материалов формируется опора, объединяющая два верхних позвонка и основание черепа.
Органы слуха.
Нейросенсорная и кондуктивная тугоухость в большинстве случаев могут компенсироваться подбором слуховых аппаратов (слуховые импланты среднего уха). При частых отитах проводится шунтирование барабанных полостей для оттока жидкости.
Сердечно-сосудистая система.
Из-за развития клапанных дисфункций нередко рекомендована консультация кардиохирурга для решения вопроса о необходимости протезирования клапанов сердца. Из-за часто развивающихся нарушений ритма ставится вопрос об имплантации кардиовертера-дефибриллятора.
Инфекции.
Важно, чтобы осуществлялся хороший уход за зубами, так как разрушенные зубы причиняют сильный дискомфорт и являются очагами инфекции. Необходимо регулярно чистить зубы, использовать растворы для полоскания. Но даже при самой тщательной заботе о зубах, могут развиваться воспалительные процессы. Раздражительность, плач и беспокойство иногда могут быть единственными симптомами проблем с зубами. Перед оперативным лечением зубов пациентам, у которых уже диагностировано поражение клапанов сердца, необходим профилактический прием антибиотиков до и после лечения. Это вызвано тем, что некоторые бактерии из ротовой полости могут проникнуть в кровоток и вызвать инфекционный процесс на пораженном клапане. Если необходимо удаление зуба под анестезией, это должно быть сделано в больнице, а не в стоматологической поликлинике, под наблюдением опытного анестезиолога и зубного врача.
Лечение банального ОРВИ у пациентов с МПС лучше также проводить под пристальным вниманием врача. Следует помнить об особенностях применения стандартных препаратов у этой группы больных. Например, такие лекарства, как R06 антигистаминные препараты, могут сушить слизистую, делая ее толще, противоотечные или сосудосуживающие средства могут повысить кровяное давление и сузить кровеносные сосуды, что нежелательно при МПС.
Из-за особенностей строения позвоночника и грудной клетки, пациентам с мукополисахаридозом I типа сложнее справиться с инфекцией, если она затрагивает легкие, поэтому врачи даже при незначительной инфекции могут назначать антибиотики.
Чтобы нормализовать циркуляцию воздуха в легких, рекомендуется и профилактически, и во время болезни проводить игры с мыльными пузырями, ватой, соломинками для напитков, бумажными пакетами и другими предметами (воздушными шарами, мячами), требующими активной работы дыхательной системы. Для детей постарше, для подростков можно разработать упражнения для растяжения мышц грудной клетки, чтобы родители могли самостоятельно заниматься с детьми.
Пассивные упражнения для грудной клетки применяются для больных с острыми инфекциями дыхательных путей или для пациентов, которые не могут самостоятельно двигаться. Упражнения помогают более эффективному отделению слизи. К ним относятся потягивания, растяжения, потряхивания, массаж и вибрации.
Диетотерапия.
Специальная диета не сможет предотвратить накопление ГАГ, так как они синтезируются в клетках, а не поступают с пищей. Поэтому ограничения в диете пациентам с МПС не нужны, дети должны питаться в соответствии с возрастными потребностями.
Патогенетическая терапия.
Для МПС тип I разработана специальная ферментная заместительная терапия.
Эффект от терапии во многом зависит от того, когда было начато лечение. Вместе с врачом необходимо перед началом ферментной заместительной терапии обсудить все возможные риски, побочные эффекты и главное - ожидания от лечения. Важно понимать, что введение фермента не позволит вылечить заболевание, цель терапии - замедлить прогрессию болезни. Те деформации, которые уже сформировались, терапия не исправит, инфузионная терапия не заменит необходимые хирургические вмешательства.
Трансплантация костного мозга/гемопоэтических стволовых клеток.
Рекомендовано проведение ТГСК пациентам с синдромом Гурлер до достижения возраста двух лет при нормальных или субнормальных показателях развития интеллекта (DQ>70). Одним из наиболее значимых эффектов трансплантации является сохранение интеллектуального развития детей, имеющих тяжелый генотип, ассоциированный с серьезными умственными расстройствами. У этих пациентов были отмечены существенные изменения черт лица (уменьшения проявлений гаргоилизма ), улучшение или стабилизация сердечно-сосудистой системы. Однако изменения скелета сохранились, в некоторых случаях отмечалось даже ухудшение ситуации. У многих пациентов наблюдалось улучшение электроретинограммы (функция сетчатки) через 1-2 года после пересадки костного мозга, однако последующие наблюдения показали, что с течением времени происходит ухудшение. Не было получено однозначных данных о влиянии пересадки костного мозга на степень помутнения роговицы.
Нельзя не отметить, что сама процедура проведения трансплантации довольно рискованна для пациента, в связи с частыми осложнениями, вызванными «отторжением» трансплантанта организмом больного.
Половая зрелость и брак.
Подростки с синдромом Гурлер-Шейе и Шейе проходят нормальные стадии полового созревания, однако начинается оно несколько позже.
Таблица 1 - План обследования пациентов с МПС I.
| Исследования | Первоначальная оценка | Каждые 3 мес | Каждые 6 мес | Каждые 12 мес | Один раз в 2 года |
| Прием врача-генетика | Х | Х | |||
| Подтверждение диагноза | Х | ||||
| Анамнез болезни | Х | ||||
| Клинический осмотр терапевта | Х | Х | |||
| Рост, вес, окружность головы | Х | Х | |||
| Выносливость (6-ти минутный тест ходьбы) | Х | Х | |||
| Осмотр невролога | Х | Х | |||
| Осмотр врача-нейрохирурга | Х | Х | |||
| Компьютерная томография или МРТ головного мозга | Х | Х | |||
| Когнитивное тестирование (DQ/IQ) | Х | Х | |||
| Электроэнцефалограмма | Х | Х | |||
| Скорость нервной проводимости/ ЭМГ | Х | Х | |||
| Осмотр врача-оториноларинголога | Х | Х | |||
| Осмотр сурдолога | Х | Х | |||
| Аудиометрия | Х | Х | |||
| Осмотр офтальмолога | Х | Х | |||
| Острота зрения | Х | Х | |||
| Осмотр глазного дна | Х | Х | |||
| Исследование роговицы | Х | Х | |||
| Осмотр пульмонолога | Х | Х | |||
| ФВД | Х | Х | |||
| Исследование сна/полисомнография | Х | Х | |||
| Осмотр кардиолога | Х | Х | |||
| Электрокардиограмма | Х | Х | |||
| Эхокардиография | Х | Х | |||
| Осмотр ортопеда | Х | Х | |||
| Рентгенографии скелета | Х | Х | |||
| УЗИ органов брюшной полости; объем селезенки, печени | Х | Х | |||
| Осмотр врача-физиотерапевта | Х | Х | |||
| Прием врача по лечебной физкультуре | Х | Х | |||
| Осмотр врача-стоматолога | Х | Х | |||
| Лабораторные исследования | |||||
| Определение ГАГ | Х | Х | |||
| Биохимический анализ крови (аланинаминотрансфераза (АЛТ), аспартатаминотрансфераза (АСТ), общий и прямой билирубин, холестерин, триглицериды) | Х | Х | |||
Заболевание не влияет на фертильность, но женщинам с тяжелой формой заболевания иногда сложно выносить ребенка в связи с тяжелыми нарушениями со стороны сердца и скелета. В мире и в России есть семьи, матери которых родили несколько здоровых детей. Поэтому все вопросы, связанные с беременностью и родами нужно решать индивидуально.
Все дети от родителей с синдромом Гурелр-Шейе и Шейе являются носителями заболевания, но не болеют, если второй родитель здоров.
Отдых.
Пациентам с мягкой формой заболевания необходимо научиться быть независимыми от своих семей. Не давайте ребенку, подростку замкнуться в себе. Ему нужны друзья, общение и навыки самостоятельной жизни.
Поскольку дети с мягкой формой синдрома имеют нормальный интеллект, необходимо приложить все усилия, чтобы они получили хорошее образование.
В каждом из регионов определены лечебные учреждения, в которых пациенты получают и патогенетическое лечение и проходят все необходимые исследования.
Узнать больше.
В мире есть много организаций, которые могут помочь вам найти ответы на любые вопросы, связанных с мукополисахаридозами:
Международная организация по мукополисахаридозам: www. mpssociety.org.
Международный портал по редким болезням www.orpha.net.
Европейская организация, объединяющая пациентов с разными редкими заболеваниями EURORDIS www.eurordis.org.
Помните, что активная позиция самих пациентов - залог успешной и счастливой жизни.
Общественные организации и фонды России.
МПС I типа входит в перечень орфанных заболеваний, лечение которых проводится за счёт средств государства. Помочь пациентам получить лечение, повысить информированность общества о редких болезнях, а также добиваться продвижения законодательных инициатив в области орфанных заболеваний могут общественные организации. В сети интернет вы сможете найти информацию о нескольких общественных организациях, которые помогают семьям с мукополисахаридозами.
Приложение Г1-ГN.
Шкалы оценки, вопросники и другие оценочные инструменты состояния пациента, приведенные в клинических рекомендациях.Приложение Г1.
Классификация мукополисахаридозов.Классификация мукополисахаридозов.
| МПС | Подтип | Синдром | Ген | Ферментный дефект | Локус | OMIM |
| I | МПС I H | Гурлер | IDUA | Дефицит альфа-L-идуронидазы | 4p16,3 | 607014 |
| МПС I H/S | Гурлер-Шейе | 607015 | ||||
| МПС I S | Шейе | 607016 | ||||
| II | МПС II | Хантера | IDS | Дефицит идуронат-2-сульфатазы | Xq28 | 309900 |
| III | МПС IIIА | Санфилиппо | SGSH | Дефицит гепаран-N-сульфатазы | 17q25,3 | 252900 |
| МПС IIIВ | NAGLU | Дефицит N-ацетил-α-D-глюкозаминидазы Дефицит N-ацетил-α-глюкозаминидазы | 17q21,2 | 252920 | ||
| МПС IIIС | HGSNAT | Дефицит гепаран-α-глюкозаминид N-ацетилтрансферазы | 8p11,21 | 252930 | ||
| МПС IIID | GNS | Дефицит N-ацетилглюкозамин-6- сульфатазы | 12q14 | 252940 | ||
| IV | МПС IVА | Моркио | GALNS | Дефицит галактозамин-6-сульфатазы | 16q24/3 | 253000 |
| МПС IVВ | GLB1 | Дефицит β-галактозидазы | 3p21,33 | 253010 | ||
| VI | МПС VI | Марото-Лами | ARSB | Дефицит N-ацетилгалактозамин-4-сульфатазы | 5q14,1 | 253200 |
| VII | МПС VII | Слая | GUSB | Дефицит β-глюкуронидазы | 7q21,11 | 253220 |
| IX | МПС IX | Недостаточность гиалуронидазы | HYAL1 | Дефицит гиалуронидазы | 3p21,31 | 601492 |
Приложение Г2.
Выраженность клинических проявлений МПС I в разном возрасте.Выраженность клинических проявлений МПС I в разном возрасте.
| Система | Симптом | Дети | Подростки/ взрослые |
| Сердечно-сосудистая | Кардиомиопатия | + | + |
| Утолщение створок клапанов | +++ | +++ | |
| ЦНС | Нарушение поведения | ++ | ++ |
| Цервикальная миелопатия | ++ | ++ | |
| Гидроцефалия | ++ | ++ | |
| Задержка и отставание в развитии | +++ | ++ | |
| Судороги | + | ++ | |
| Нарушение глотания | + | ± | |
| Пищеварительная система | Диарея | + | ± |
| Гепатоспленомегалия | +++ | +++ | |
| Орган слуха | Потеря слуха | ++ | ++ |
| Рецидивирующий средний отит | ++ | ++ | |
| Орган зрения | Дистрофия сетчатки | ± | ± |
| Помутнение роговицы | + | +++ | |
| Скелетно- мышечная | Нестабильность атланто-окципитального сочленения | + | + |
| Туннельный карпальный синдром | ++ | ++ | |
| Грубые черты лица | +++ | ++ | |
| Дисплазия ТБС | ++ | ++ | |
| Множественный дизостоз | + | + | |
| Грыжи | +++ | ++ | |
| Контрактуры суставов | +++ | +++ | |
| Кифоз | ++ | + | |
| Макроцефалия | +++ | ++ | |
| Дыхательная | Обструктивное апноэ во сне | +++ | +++ |
| Рестриктивное заболевание легких | + | + | |
| Обструкция верхних дыхательных путей | +++ | +++ | |
| Специаль- ные лабораторные тесты | Дерматан сульфат в моче | ↑↑ | ↑↑ |
| Гепаран сульфат в моче | ↑↑ | ↑↑ | |
| Идуронидаза | ↓↓↓ | ↓↓↓ | |
| Общие ГАГ в моче | ↑↑ | ↑↑ |
Приложение Г3 Частота проведения обследования у пациентов с МПС I типа.
| Исследования | Первоначальная оценка | Каждые 3 мес | Каждые 6 мес | Каждые 12 мес | Один раз в 2 года |
| Прием врача-генетика | Х | Х | |||
| Подтверждение диагноза | Х | ||||
| Анамнез болезни | Х | ||||
| Клинический осмотр терапевта | Х | Х | |||
| Рост, вес, окружность головы | Х | Х | |||
| Выносливость1 | Х | Х | |||
| Осмотр невролога | Х | Х | |||
| Осмотр врача-нейрохирурга | Х | Х | |||
| Компьютерная томография или МРТ головного мозга | Х | Х | |||
| Когнитивное тестирование (DQ/IQ)2 | Х | Х | |||
| Электроэнцефалограмма | Х | Х | |||
| Скорость нервной проводимости/ ЭМГ | Х | Х | |||
| Осмотр врача-оториноларинголога | Х | Х | |||
| Осмотр сурдолога | Х | Х | |||
| Аудиометрия | Х | Х | |||
| Осмотр офтальмолога | Х | Х | |||
| Острота зрения | Х | Х | |||
| Осмотр глазного дна | Х | Х | |||
| Исследование роговицы | Х | Х | |||
| Осмотр пульмонолога | Х | Х | |||
| ФВД | Х | Х | |||
| Исследование сна/полисомнография | Х | Х | |||
| Осмотр кардиолога | Х | Х | |||
| Электрокардиограмма | Х | Х | |||
| Эхокардиография | Х | Х | |||
| Осмотр ортопеда | Х | Х | |||
| Рентгенографии скелета | Х | Х | |||
| УЗИ органов брюшной полости; объем селезенки, печени | Х | Х | |||
| Осмотр врача-физиотерапевта | Х | Х | |||
| Прием врача по лечебной физкультуре | Х | Х | |||
| Осмотр врача-стоматолога | Х | Х | |||
| Лабораторные исследования | |||||
| Определение ГАГ | Х | Х | |||
| Биохимический анализ крови | Х | Х | |||
1-Для детей старше 3 лет; расстояние, пройденное за 6 минут (предпочтительным является тот же промежуток времени, что и в предыдущих тестах этого пациента); число ступеней, пройденных вверх за 3 минуты. Оцениваются показатели сатурации SpO2, ЧДД, ЧСС до и после нагрузки.
2-DQ или Коэффициент умственного развития (КУР) - способ сравнения интеллектуального развития, свойственного данному возрасту (умственных способностей ребенка по отношению к его сверстникам), с хронологическим возрастом (фактическим возрастом ребенка).
КУР рассчитывается делением интеллектуального возраста на хронологический и умножением на 100 для получения целого числа. Средний коэффициент умственного развития для любого возраста считается равным 100.
Приложение Г4.
Тест 6 минутной ходьбы (6MWT).Название на русском языке: тест 6-минутной ходьбы.
Оригинальное название (если есть): The six minute walking test (6MWT).
Источник (официальный сайт разработчиков, публикация с валидацией): Laboratories, A. T. S. с. o. P. S. f. с. P. F. (2002). ATS statement: guidelines for the six-minute walk test. Am J Respir сrit сare Med 166(1): 111-117.
Тип (подчеркнуть):
Шкала оценки.
Индекс.
Вопросник.
Другое.
Назначение: определение выносливости пациентов с целью оценки эффективности терапии.
Содержание (шаблон): в тесте оценивается дистанция в метрах, пройденная пациентом за 6 минут без вынужденных остановок.
Ключ (интерпретация): проводится сравнение с результатами теста, проведенного ранее (до начала терапии, на фоне терапии и тд).
6MWT. Тест 6 - минутной ходьбы (6 minutes walking test, 6MWT).
Следует помнить, что для данного теста имеются следующие абсолютные противопоказания: нестабильная стенокардия напряжения и инфаркт миокарда, давностью до 1 месяца. Относительными противопоказаниями являются: ЧСС выше 120/мин в покое, систолическое АД >180 и диастолическое АД better 100 Стабильная стенокардия не является абсолютным противопоказанием для проведения теста, однако его следует проводить с осторожностью, на фоне приема антиангинальных препаратов по показаниям.
Если пациент находится на постоянной кислородной поддержке, скорость подачи кислорода при проведении теста должна сохраняться в соответствии с предписанием врача, назначившего и контролирующего терапию.
Проведение теста необходимо немедленно прекратить в случае появления:
• Боли в груди;
• Непереносимой одышки;
• Крампи в ногах;
• Резкой неустойчивости и пошатывания при ходьбе;
• Чрезмерного потоотделения;
• Резкого изменения цвета кожных покровов (бледности).
6MWT проводится в помещении, хорошо знакомом пациенту, имеющем достаточно длинный коридор с твердым покрытием. Длина проходимого пути должна составлять не менее 30 метров с разметкой каждые 3 метра, а также точками поворотов/разворотов.
Пациент получает инструкцию о необходимости идти с максимально возможной скоростью (но не бежать) в течение 6 минут.
В тесте оценивается дистанция в метрах, пройденная пациентом за 6 минут без вынужденных остановок.
Приложение Г5.
Забор биоматериала для диагностики в пятнах крови.| Кровь собирается на стандартную карточку-фильтр (№903), которая применяется для скрининга новорожденных в Российской Федерации или аналогичную для получения сухого пятна крови. Кровь может быть, как капиллярная (из пальца, пятки), так и венозная. Венозная кровь собирается в пробирку, аккуратно перемешивается и затем быстро с помощью пипетки наносится на фильтр по 25-50 мкл крови на каждую выделенную область. Необходимо хорошо пропитать выделенную область на фильтре ( 1). Предпочтительно собирать образцы после еды через 40 минут - 1 час. Возможно также осуществить забор крови и натощак. На карточке-фильтре обязательно должны быть четко указаны ФИО, кем и откуда направлен пациент, дата рождения и телефон лечащего врача (рисунок 2). Образец сухого пятна крови вкладывается в чистый конверт, либо в чистый файл. Карточка-фильтр не должна соприкасаться с грязной поверхностью и с образцами других пациентов. Необходимо приложить к образцам информированные согласия пациента или его законных представителей на проведение лабораторных исследований | Рисунок 1. Образец правильного нанесения крови на карточку-фильтр Рисунок 2. Образец карточки-фильтра |
Алгоритм действий медицинского персонала при взятии образцов крови.
• вымыть руки (гигиенический уровень), надеть перчатки;
• вымыть руки пациента (пятку ребенка, в случае, если кровь берется из пятки);
• протереть область прокалывания стерильной салфеткой, смоченной 70% спиртом, промокнуть сухой стерильной салфеткой; - проколоть стерильным одноразовым скарификатором;
• снять первую каплю крови стерильным сухим тампоном;
• мягко надавить для получения второй капли крови;
• приложить перпендикулярно тест-бланк к капле крови и пропитать его кровью насквозь;
• аналогичным образом нанести на тест-бланк 6-8 капель, вид пятен крови должен быть одинаковым с обеих сторон.
• высушить тест-бланк в горизонтальном положении на чистой обезжиренной поверхности не менее 4 ч без применения тепловой обработки и попадания прямых солнечных лучей;
• упаковать тест-бланки в чистый конверт таким образом, чтобы пятна крови не соприкасались.
Особенности при инфузионной терапии.
Некоторые пациенты могут получать инфузионную терапию, переливание компонентов крови, что может оказать влияние на результаты тестов. Например, при переливании плазмы крови могут быть получены ложно-отрицательные результаты, так как определяемые ферменты находятся в плазме и в клетках крови. Рекомендуется осуществить забор крови для ферментной и ДНК-диагностики не 9 ранее чем через 6-7 дней после переливания плазмы крови и через 7-10 дней после переливания компонентов крови.
Не допускается забор крови.
• сразу после проведения пациенту инфузионной терапии;
• сразу после заменного переливания крови.
Хранение и транспортировка биоматериала.
Образцы высушенных пятен крови можно хранить в обычной камере холодильника при +40С до отправки. Срок хранения до момента отправки не должен превышать 7 дней. Если хранить дольше и при более высокой температуре, то активность фермента даже в норме может снижаться, что приведет к ложно-положительным результатам.
|
|
Год актуализации информации
2019.
Связанные заболевания
Связанные клинические рекомендации
Связанные стандарты мед. помощи
- Стандарт медицинской помощи взрослым при мукополисахаридозе I типа
- Стандарт специализированной медицинской помощи детям при мукополисахаридозе I типа (ферментная заместительная терапия)
- Стандарт специализированной медицинской помощи детям при мукополисахаридозе I типа (диагностика и инициация ферментной заместительной терапии)
- Стандарт первичной медико-санитарной помощи детям при мукополисахаридозе I типа (ферментная заместительная терапия)