Другие названия и синонимы
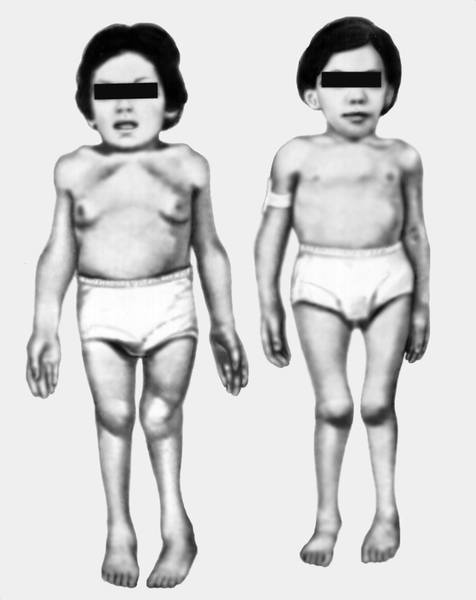
Morchio 's disease.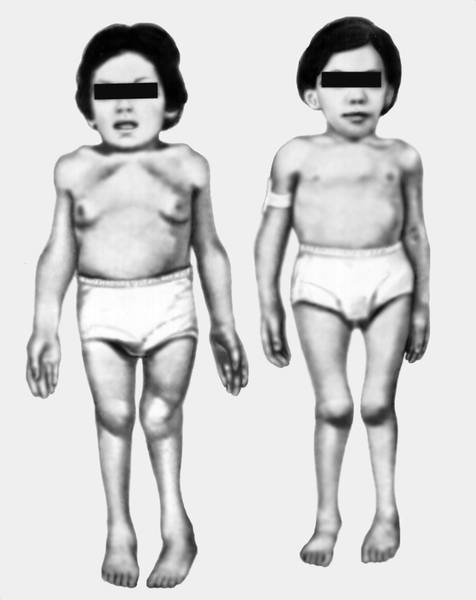
МКБ-10 коды
|
|
Описание
Болезнь Моркио ( описана уругвайским педиатром L. Morquio, 1867-1935) относится к мукополисахаридозам (IV тип), то есть к группе наследственных болезней соединительной ткани, обусловленных нарушением обмена гликозаминогликанов (кислых мукополисахаридов) в результате генетически обусловленной неполноценности ферментов, участвующих в их расщеплении и которые наследуется по аутосомно-рецессивному типу. При мукополисахаридозах поражается система лизосомных ферментов, участвующих в катаболизме гликозаминогликанов. Вследствие ферментативной недостаточности последние накапливаются в большом количестве в органах и тканях, поэтому мукополисахаридозы относят к болезням накопления. В результате нарушается функциональное состояние различных органов и систем. Поскольку гликозаминогликаны входят в состав соединительной ткани, то одним из ведущих проявлений мукополисахаридоза является системное поражение скелета, задержка физического развития.
Причины
Болезнь Моркио передается по аутосомно-рецессивному типу. Частота до 1: 40 000. Дети рождаются без признаков болезни. Первые симптомы появляются в возрасте 1-3 года, и к 7-8 годам клиническая картина уже полностью выражена.
Дефицитарный фермент - галакто-бета-сульфат-сульфатаза (тип А); бета-галактозидаза (тип В).
Дефицитарный фермент - галакто-бета-сульфат-сульфатаза (тип А); бета-галактозидаза (тип В).
Клиническая картина
Болезнь характеризуется карликовостью (рост взрослого больного около 100 см), непропорциональным телосложением (относительно короткое туловище, микроцефалия, короткая шея), грубыми чертами лица и значительными деформациями скелета, особенно грудной клетки (куриная, бочкообразная, килеобразная), кифозом или сколиозом грудного и поясничного отделов позвоночника. Питание снижено. Тугоподвижность в суставах и вместе с тем расслабление сумочно-связочного аппарата в мелких суставах, шея укорочена, гипоплазия отростков I и II шейных позвонков. В случае компрессии спинного мозга помимо мышечной гипотонии отмечается поражение пирамидной системы, возможно развитие параплегии, паралича дыхания. Возникают контрактуры в локтевых, плечевых, коленных суставах, отмечается вальгусная деформация нижних конечностей, плоскостопие. Интеллект не нарушен, отсутствует гепатоспленомегалия. Лицо обычное, размеры черепа без изменений. Кожа утолщена, ее тургор и эластичность снижены. Часто выявляются пупочные и паховые грыжи, расхождение прямых мышц живота. Иногда отмечается нерезко выраженное помутнение роговиц. Нередко отмечается снижение слуха. Почти у всех больных, доживших до 20 лет, развивается глухота. В поздний срок болезни появляются нарушения сердечнососудистой системы, истончение зубов. Кроме того у больных могут наблюдаться кардиопатия и миопатия. Как правило, мышечная сила снижена. С мочой выделяется большое количество кератансульфата. В большинстве случаев выраженных клинических проявлений летальный исход наступает до 20 лет вследствие сердечно-легочной недостаточности, развивающейся на фоне интеркуррентных заболеваний. Возможна внезапная смерть в результате смещения атланто-окципитального сочленения и повреждения ствола мозга.
Диагностика
Рентгенологически видны характерные изменения в позвоночном столбе: уплощение тел позвонков, снижение высоты их передних отделов, запаздывание в развитии апофизов, особенно в XII грудном и I поясничном позвонках. Кифосколиоз. Во всех отделах отмечается платиспондилия - уплощение и расширение тел позвонков, чем объясняется характерное укорочение туловища и необычно короткая шея. Эпифизы длинных трубчатых костей недоразвиты. Окостенение метафизов также нерегулярное и позднее. Кисти укорочены, широкие пальцы утолщены, с конической формой эпифизов. Кости предплечья укорочены; локтевая кость не достигает лучезапястного сустава, отмечается вывих ее головки в локтевом суставе; эпифизы треугольной формы. Дистальные эпифизы костей голени скошены, стопы деформированы. Изменяются кости таза: вертлужные впадины плоские и широкие, их крыша скошена, крылья подвздошных костей неправильной формы; контуры всех костей неровные; головки бедренных костей уплощены. Грибовидная головка бедренной кости внедряется в углубленную вертлужную впадину. шейка бедра укорачивается. поражение суставов двустороннее, в результате чего уменьшается поперечник таза. Нередки остеоартриты, особенно в тазобедренных суставах. Характерна увеличивающаяся с возрастом вальгусная деформация нижних конечностей. Уплощение эопифизов бедренной и большеберцовой кости, сглаживание межмыщелковых бугорков сопровождаются специфической деформацией коленных суставов. Поражение голеностопного сустава обуславливает неправильную установку стопы.
Диагноз ставится на основании характерных внешних проявлений болезни. В качестве биохимического скрининга применяют исследования экскреции с мочой метаболитов мукополисахаридов и активности лизосомальных ферментов в лейкоцитах крови и культуре фибробластов кожи. Диагнозу может помочь семейный анамнез. При постановки диагноза следует иметь в виду, что различные биохимические дефекты могут приводить к сходным клиническим симптомам и наоборот, недостаток одного и тогоже фермента может иметь разные фенотипические проявления. Диагноз подтверждается оценкой активности лизосомальных ферментов в плазме или лейкоцитах крови, культуре фибробластов кожи и, в ряде случаев, исследованием ДНК. Возможен пренатальный диагноз.
Диагноз ставится на основании характерных внешних проявлений болезни. В качестве биохимического скрининга применяют исследования экскреции с мочой метаболитов мукополисахаридов и активности лизосомальных ферментов в лейкоцитах крови и культуре фибробластов кожи. Диагнозу может помочь семейный анамнез. При постановки диагноза следует иметь в виду, что различные биохимические дефекты могут приводить к сходным клиническим симптомам и наоборот, недостаток одного и тогоже фермента может иметь разные фенотипические проявления. Диагноз подтверждается оценкой активности лизосомальных ферментов в плазме или лейкоцитах крови, культуре фибробластов кожи и, в ряде случаев, исследованием ДНК. Возможен пренатальный диагноз.
Лечение
Как и при других лизосомальных болезнях накопления, эффективного лечения неврологических осложнений мукополисахаридозов нет. При развитии гидроцефалии, сдавлении спинного мозга, нестабильности атланто-аксиального сочленения и туннельных невропатий показано хирургическое вмешательство. Перспективный метод лечения мукополисахаридозов - генная терапия. Очень важно симптоматическое лечение.
|
|