Другие названия и синонимы
Gurler syndrome, Болезнь Пфаундлера-Гурлер, Мукополисахаридоз IH.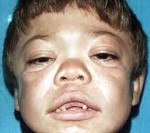
МКБ-10 коды
|
|
Описание
Это тяжелое наследственное нарушение обмена веществ из группы мукополисахаридозов, характеризующееся чрезмерным накоплением гликозаминогликанов (ГАГ) в различных органах и тканях, что приводит к их выраженной дисфункции. Клиническая картина чрезвычайно разнообразна, включая задержку психомоторного развития, тяжелые деформации костей черепа и скелета, сердечно-легочные расстройства Диагноз основан на определении экскреции гликозаминогликана с мочой и активности фермента альфа-идуронидазы в крови, данных молекулярно-генетических тестов. Лечение состоит из фермент-заместительной терапии.
Дополнительные факты
Синдром Hurler (болезнь Pfoundler-Hurler, мукополисахаридоз IH или первый тип) относится к лизосомным болезням накопления с типом аутосомно-рецессивной передачи, проявляющейся почти в первые месяцы жизни. Болезнь была впервые описана австрийским педиатром Гертрудой Гурлер. Патология считается одним из наиболее распространенных мукополисахаридозов, встречающихся повсеместно. По разным данным, синдром Херлера выявляется у новорожденных в 1:40 000-1: 100 000. Существенных статистических различий между полами нет.
Причины
Возникновение заболевания связано с гетерогенными мутациями (небольшие делеции, дефекты в месте соединения) гена IDUA, который кодирует синтез фермента альфа-L-идуронидазы. Ген расположен в коротком плече хромосомы 4 в локусе 4p16.3. Наиболее распространенными генными мутациями являются Q70X и W402X. Лизосомный фермент α-L-идуронидаза регулирует метаболизм основных структурных компонентов межклеточного матрикса соединительной ткани - он отвечает за деградацию гепарансульфата и дерматансульфата гликозаминогликанов.
Из-за генетически детерминированного дефекта фермента происходит чрезмерное накопление GAG в клеточных лизосомах. Наиболее значимым фактором риска развития синдрома Херлера является наличие близкого родственника, пораженного этим заболеванием. Если один из родителей имеет мутантный ген, вероятность рождения больного ребенка составляет 25%.
Из-за генетически детерминированного дефекта фермента происходит чрезмерное накопление GAG в клеточных лизосомах. Наиболее значимым фактором риска развития синдрома Херлера является наличие близкого родственника, пораженного этим заболеванием. Если один из родителей имеет мутантный ген, вероятность рождения больного ребенка составляет 25%.
Патогенез
В результате нарушения лизосомального гидролиза с последующим накоплением межклеточных компонентов соединительной ткани высвобождаются медиаторы воспаления - развивается оксид азота, фактор альфа некроза опухоли и дисфункция органов. Поскольку соединительная ткань, в той или иной степени, является частью почти каждого органа, изменение является мультисистемным.
Накопление мукополисахаридов в хряще вызывает нарушение роста костей, их выраженную деформацию. Фиброз развивается в стенках кровеносных сосудов, клапанном аппарате и мозговых оболочках. Раздел экспертизы выявляет расширение органа, гиперцеллюлярность и дезорганизацию. Отмечено уменьшение количества протеогликанов и коллагеновых волокон.
Накопление мукополисахаридов в хряще вызывает нарушение роста костей, их выраженную деформацию. Фиброз развивается в стенках кровеносных сосудов, клапанном аппарате и мозговых оболочках. Раздел экспертизы выявляет расширение органа, гиперцеллюлярность и дезорганизацию. Отмечено уменьшение количества протеогликанов и коллагеновых волокон.
Клиническая картина
Клинические признаки заболевания появляются в первый месяц жизни. Иногда увеличение селезенки и печени, пупочных и паховых грыж отмечается с рождения. Симптомы повреждения опорно-двигательного аппарата произносятся. Контрактуры и тугоподвижность суставов развиваются достаточно быстро, образуются патологические изгибы позвоночника (кифоз поясничного отдела позвоночника).
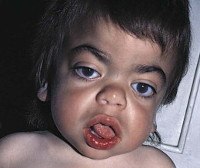
В конце первого года жизни на лице ребенка проявляются характерные черты типа «горгулизм» - выступающие лобные бугорки, широкий уплощенный нос, гипертелоризм глаз, толстые губы и язык, суженная часть лица черепа. Типичная задержка роста, которая почти полностью прекращается через 4-5 лет. По мере прогрессирования заболевания появляются симптомы повреждения центральной и периферической нервной системы.
Наблюдается заметная задержка психомоторного развития. Интеллект снижен, язык не развит. Дефекты речи также способствуют формированию нейросенсорной тугоухости. Выделение и агрессия отмечены при поведенческих расстройствах. Походка нарушена, тонус мышц снижен. Иногда возникают судороги вплоть до тонико-клонических пароксизмов.
Другие признаки синдрома Херлера включают распространенные инфекционные заболевания верхних дыхательных путей, рецидивирующий средний отит и непрозрачность роговицы. Из-за накопления ГАГ в миндалинах, трахее и надгортаннике, просвет дыхательной трубки постепенно сужается, что увеличивает риск обструктивного апноэ во сне.
Ассоциированные симптомы: Судороги в ногах.
В конце первого года жизни на лице ребенка проявляются характерные черты типа «горгулизм» - выступающие лобные бугорки, широкий уплощенный нос, гипертелоризм глаз, толстые губы и язык, суженная часть лица черепа. Типичная задержка роста, которая почти полностью прекращается через 4-5 лет. По мере прогрессирования заболевания появляются симптомы повреждения центральной и периферической нервной системы.
Наблюдается заметная задержка психомоторного развития. Интеллект снижен, язык не развит. Дефекты речи также способствуют формированию нейросенсорной тугоухости. Выделение и агрессия отмечены при поведенческих расстройствах. Походка нарушена, тонус мышц снижен. Иногда возникают судороги вплоть до тонико-клонических пароксизмов.
Другие признаки синдрома Херлера включают распространенные инфекционные заболевания верхних дыхательных путей, рецидивирующий средний отит и непрозрачность роговицы. Из-за накопления ГАГ в миндалинах, трахее и надгортаннике, просвет дыхательной трубки постепенно сужается, что увеличивает риск обструктивного апноэ во сне.
Ассоциированные симптомы: Судороги в ногах.
Возможные осложнения
Синдром Херлера - серьезное заболевание со многими осложнениями. Основными причинами смерти считаются прогрессирующая хроническая сердечная недостаточность из-за возникающих заболеваний сердца и кардиомиопатии, дыхательная недостаточность из-за обструкции дыхательных путей и тяжелые инфекции (пневмония, менингит, туберкулез).
Гидроцефалия может привести к отеку мозга. Сердечные аритмии возникают из-за фиброзного повреждения миокарда. Сжатие спинного мозга, квадриплегия или параплегия может привести к ухудшению или полной потере тазовых функций. Иногда наблюдается потеря зрения и слуха.
Гидроцефалия может привести к отеку мозга. Сердечные аритмии возникают из-за фиброзного повреждения миокарда. Сжатие спинного мозга, квадриплегия или параплегия может привести к ухудшению или полной потере тазовых функций. Иногда наблюдается потеря зрения и слуха.
Диагностика
Из-за полиорганного повреждения пациенты с синдромом Херлера нуждаются в междисциплинарном подходе, поэтому наблюдение осуществляется врачами различных специальностей - педиатрами, неврологами и кардиохирургами. Анамнестические данные и яркие фенотипические признаки заболевания помогают заподозрить наличие патологии. Для подтверждения диагноза рекомендуется дополнительное обследование, в том числе:
• Определение ферментативной активности. Снижение лизосомальной активности α-L-идуронидазы наблюдается в лейкоцитах периферической крови.
• Выведение GAG с мочой. Повышенное содержание гликозаминогликанов в моче было обнаружено из-за дерматансульфата и гепарансульфата.
• МРТ головного и спинного мозга. На МРТ головного мозга наблюдается утолщение мозговых оболочек, признаки гидроцефалии (вазодилатация, цистерны), на МРТ позвоночника - сдавление спинного мозга.
Рентгенологические признаки синдрома Херлера включают расширение диафиза трубчатых костей, укорочение и утолщение ключицы и «ребристую» форму ребер. Рентген позвоночника указывает на его искривление, расширение позвонков, недоразвитие, деформацию поперечных отростков.
Эхокардиография показывает утолщение или деформацию створок клапана, признаки регургитации, расширение камер сердца. С развитием сердечной недостаточности фракция выброса левого желудочка уменьшается.
Во время УЗИ или КТ органов брюшной полости определяется диффузное увеличение печени и селезенки.
При измерении функции внешнего дыхания выявляются обструктивные нарушения - снижение форсированной жизненной емкости легких, максимальный инспираторный поток.
У некоторых пациентов с компрессией нервного ствола при электронейромиографии наблюдаются признаки невропатии - закупорка и замедление движения нервного импульса.
Выполнение аудиометрии показывает значительное увеличение порога восприятия звука. Из-за раннего возраста пациентов предпочтительны объективные методы (компьютерной) аудиометрии.
Очень часто при исследовании глазного дна, отека и застоя зрительного диска определяется периферическая дистрофия сетчатки.
Молекулярно-генетическое исследование для выявления мутаций в гене IDUA считается решающим в диагностике заболевания.
Дифференциальный диагноз синдрома Херлера ставится при других наследственных нарушениях обмена веществ - II, III типах мукополисахаридоза, ганглиозидоза, множественной сульфатазной недостаточности. Повреждение суставов следует отличать от неинфекционного артрита и ювенильного ревматоидного артрита.
• Определение ферментативной активности. Снижение лизосомальной активности α-L-идуронидазы наблюдается в лейкоцитах периферической крови.
• Выведение GAG с мочой. Повышенное содержание гликозаминогликанов в моче было обнаружено из-за дерматансульфата и гепарансульфата.
• МРТ головного и спинного мозга. На МРТ головного мозга наблюдается утолщение мозговых оболочек, признаки гидроцефалии (вазодилатация, цистерны), на МРТ позвоночника - сдавление спинного мозга.
Рентгенологические признаки синдрома Херлера включают расширение диафиза трубчатых костей, укорочение и утолщение ключицы и «ребристую» форму ребер. Рентген позвоночника указывает на его искривление, расширение позвонков, недоразвитие, деформацию поперечных отростков.
Эхокардиография показывает утолщение или деформацию створок клапана, признаки регургитации, расширение камер сердца. С развитием сердечной недостаточности фракция выброса левого желудочка уменьшается.
Во время УЗИ или КТ органов брюшной полости определяется диффузное увеличение печени и селезенки.
При измерении функции внешнего дыхания выявляются обструктивные нарушения - снижение форсированной жизненной емкости легких, максимальный инспираторный поток.
У некоторых пациентов с компрессией нервного ствола при электронейромиографии наблюдаются признаки невропатии - закупорка и замедление движения нервного импульса.
Выполнение аудиометрии показывает значительное увеличение порога восприятия звука. Из-за раннего возраста пациентов предпочтительны объективные методы (компьютерной) аудиометрии.
Очень часто при исследовании глазного дна, отека и застоя зрительного диска определяется периферическая дистрофия сетчатки.
Молекулярно-генетическое исследование для выявления мутаций в гене IDUA считается решающим в диагностике заболевания.
Дифференциальный диагноз синдрома Херлера ставится при других наследственных нарушениях обмена веществ - II, III типах мукополисахаридоза, ганглиозидоза, множественной сульфатазной недостаточности. Повреждение суставов следует отличать от неинфекционного артрита и ювенильного ревматоидного артрита.
|
|
Лечение
Пациенты подлежат обязательной госпитализации. Основным лекарственным патогенетическим лечением является ферменто-заместительная терапия в течение всей жизни (ФЗТ). Используется рекомбинантная форма человеческой альфа-идуронидазы (ларонидазы). Введение этого препарата помогает восстановить ферментативную активность, достаточную для гидролиза накопленных ГАГ и предотвращения их дальнейшего осаждения.
Раннее применение ФЗТ может замедлить прогрессирование неврологических расстройств и сердечной недостаточности, восстановить активные движения в суставах и позвоночнике, добиться регресса гепатоспленомегалии и исчезновения ночного апноэ. Также проводится следующая симптоматическая терапия:
Для лечения застойной сердечной недостаточности назначаются ингибиторы АПФ (периндоприл), блокаторы бета-адренергических рецепторов (бисопролол), антагонисты альдостерона (спиронолактон).
Для предотвращения приступов используются противосудорожные средства - антагонисты NMDA-рецепторов (фенитоин), стимуляторы активности ГАМК (габапентин) и регуляторы ионных каналов.
• Психотропные препараты. Чтобы исправить поведенческие расстройства, седативные и седативные средства (бензодиазепины) подбираются индивидуально вместе с психоневрологом.
Радикальное лечение рекомендуется для пациентов с синдромом Херлера в возрасте до 2 лет, чтобы избежать необходимости пожизненной энзимотерапии - трансплантации гемопоэтических стволовых клеток (HSCT). Стволовые клетки костного мозга или пуповинной крови трансплантируются от HLA-совместимых родственных доноров. HSCT предотвращает развитие когнитивных нарушений. Назначена ФЗТ и иммуносупрессивная терапия.
При наличии соответствующих указаний выполняются следующие виды операций:
• Протезирование сердечного клапана. Болезнь сердца (недостаточность или стеноз), которая приводит к значительным нарушениям гемодинамики и прогрессированию сердечной недостаточности.
• Декомпрессия спинного мозга и нервных стволов. Компрессия спинного мозга, которая вызывает сенсомоторные и тазовые нарушения.
• Вентрикулоперитонеальный шунт. Гидроцефалия высокого давления спинномозговой жидкости.
Появление паховых или пупочных грыж (если требуется травма грыжи, требуется срочная операция).
Выраженная гипертрофия миндалин, затрудняющая дыхание.
• Протезирование коленных или тазобедренных суставов. Развитие грубых деформаций и контрактур, которые полностью ограничивают активные движения.
Раннее применение ФЗТ может замедлить прогрессирование неврологических расстройств и сердечной недостаточности, восстановить активные движения в суставах и позвоночнике, добиться регресса гепатоспленомегалии и исчезновения ночного апноэ. Также проводится следующая симптоматическая терапия:
Для лечения застойной сердечной недостаточности назначаются ингибиторы АПФ (периндоприл), блокаторы бета-адренергических рецепторов (бисопролол), антагонисты альдостерона (спиронолактон).
Для предотвращения приступов используются противосудорожные средства - антагонисты NMDA-рецепторов (фенитоин), стимуляторы активности ГАМК (габапентин) и регуляторы ионных каналов.
• Психотропные препараты. Чтобы исправить поведенческие расстройства, седативные и седативные средства (бензодиазепины) подбираются индивидуально вместе с психоневрологом.
Радикальное лечение рекомендуется для пациентов с синдромом Херлера в возрасте до 2 лет, чтобы избежать необходимости пожизненной энзимотерапии - трансплантации гемопоэтических стволовых клеток (HSCT). Стволовые клетки костного мозга или пуповинной крови трансплантируются от HLA-совместимых родственных доноров. HSCT предотвращает развитие когнитивных нарушений. Назначена ФЗТ и иммуносупрессивная терапия.
При наличии соответствующих указаний выполняются следующие виды операций:
• Протезирование сердечного клапана. Болезнь сердца (недостаточность или стеноз), которая приводит к значительным нарушениям гемодинамики и прогрессированию сердечной недостаточности.
• Декомпрессия спинного мозга и нервных стволов. Компрессия спинного мозга, которая вызывает сенсомоторные и тазовые нарушения.
• Вентрикулоперитонеальный шунт. Гидроцефалия высокого давления спинномозговой жидкости.
Появление паховых или пупочных грыж (если требуется травма грыжи, требуется срочная операция).
Выраженная гипертрофия миндалин, затрудняющая дыхание.
• Протезирование коленных или тазобедренных суставов. Развитие грубых деформаций и контрактур, которые полностью ограничивают активные движения.
Реабилитация и амбулаторное лечение
Важной частью лечения является реабилитация, которая включает в себя два основных аспекта. Для восстановления движений в суставах необходимы массажи и физиотерапевтические упражнения. Психолого-педагогическая помощь в виде систематических индивидуальных занятий направлена на развитие когнитивных функций, способствует повышению интеллекта.
Прогноз
Синдром Гурлера - серьезное заболевание с высокой смертностью. Средняя продолжительность жизни пациентов без своевременной назначенной патогенетической терапии составляет 10 лет. Наиболее распространенными причинами смерти являются респираторная и сердечная недостаточность, тяжелые бактериальные инфекции.
Профилактика
Единственная эффективная профилактика развития патологии - прерывание беременности, если во время пренатальной диагностики при обследовании ворсин хориона на 8-10 неделе беременности была обнаружена низкая активность альфа-идуронидазы. Если у вас есть близкий родственник, страдающий синдромом Херлера, рекомендуется диагностика ДНК. Для предотвращения инфекционных осложнений рекомендуется вакцинация против пневмококков, менингококков, гемофильных палочек.
Список литературы
1. Болезни накопления, сопровождающиеся нарушениями нервно-психического развития. Наследственные нарушения нервно-психического развития у детей. Руководство для врачей/ под ред. Темина П.А., Казанцевой Л.З. 2001.
2. Федеральные клинические рекомендации по оказанию медицинской помощи детям с мукополисахаридозом I типа - 2013.
3. Мукополисахаридоз I типа у детей. Клинические рекомендации - 2016.
4. Нарушения процессов клеточной биоэнергетики и методы их терапевтической коррекции у детей с мукополисахаридозами/ Семячкина А.Н., Сухоруков В.С. Российский вестник перинатологии и педиатрии - 2005 - №1.
2. Федеральные клинические рекомендации по оказанию медицинской помощи детям с мукополисахаридозом I типа - 2013.
3. Мукополисахаридоз I типа у детей. Клинические рекомендации - 2016.
4. Нарушения процессов клеточной биоэнергетики и методы их терапевтической коррекции у детей с мукополисахаридозами/ Семячкина А.Н., Сухоруков В.С. Российский вестник перинатологии и педиатрии - 2005 - №1.
Связанные клинические рекомендации
Связанные стандарты мед. помощи
- Стандарт медицинской помощи взрослым при мукополисахаридозе I типа
- Стандарт специализированной медицинской помощи детям при мукополисахаридозе I типа (ферментная заместительная терапия)
- Стандарт специализированной медицинской помощи детям при мукополисахаридозе I типа (диагностика и инициация ферментной заместительной терапии)
- Стандарт первичной медико-санитарной помощи детям при мукополисахаридозе I типа (ферментная заместительная терапия)