Другие названия и синонимы
Congenital dyskeratosis.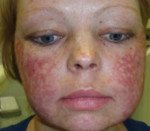
МКБ-10 коды
|
|
Описание
Врожденный дискератоз. Крайне редкое генетическое заболевание, классическая клиника которого включает локальную гиперпигментацию кожных покровов, дистрофию ногтевых пластинок и лейкоплакию. Также возникает депрессия костного мозга, что проявляется апластической анемией, иммунодефицитом и кровотечениями. Часто врожденный дискератоз осложняется злокачественными новообразованиями. Диагностика подразумевает сопоставление клинических симптомов и выявление дефектов кариотипа при помощи кариотипирования или флуоресцентной гибридизации. Лечение заключается в восстановлении нормальной функции костного мозга путем его трансплантации или фармакологической стимуляции. Параллельно проводится симптоматическая терапия.
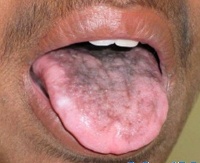
Дополнительные факты
Врожденный дискератоз - генетически обусловленный синдром, проявляющийся недостаточностью костного мозга и совокупностью специфических симптомов. Впервые классическая триада заболевания была описана в 1906 году немецким дерматовенерологом Цинссером. В 1930-х годах Коул и Энгман дополнили описание, после чего патология получила второе название - синдром Цинссера-Коула-Энгмана. На данный момент клиническая характеристика заболевания, помимо классической триады, включает другие симптомы и синдромы. Патология встречается крайне редко, распространенность - 1:1000000. Представители мужского пола болеют в 4,7 раз чаще, чем женского. Манифестация заболевания происходит в возрасте от 5 до 15 лет, поэтому первым с ним сталкивается педиатр. Симптомы развиваются постепенно, наиболее выраженная клиническая картина приходится на возраст 25-30 лет и старше. Как правило, больные умирают от осложнений в виде вторичных инфекций, злокачественных новообразований и внутренних кровотечений.
Причины
Врожденный дискератоз являет собой наследственное заболевание, которое передается по сцепленному с Х-хромосомой, аутосомно-доминантному и аутосомно-рецессивному типам. Более чем в 75% случаев передача проходит рецессивно, посредством Х-хромосомы, в 25% - по аутосомно-доминантному механизму. Локализация дефектного гена, обуславливающего развитие дискератоза - дискератина - Xq28, редко - в 3q14. Могут наблюдаться разрывы хроматид в локусах 2q33 и 8q22, аномалии митоза. Причины и механизмы развития первичного дефекта кариотипа неизвестны.
Клиническая картина
Классический вариант врожденного дискератоза включает в себя триаду симптомов: дистрофические изменения ногтей, гиперпигментацию кожных покровов и лейкоплакию слизистых оболочек. Более чем в 85% случаев его течение осложняется другими патологическими состояниями: апластической анемией, иммунодефицитом, аномалией развития мозжечка, умственной отсталостью, фиброзным перерождением легких или печени, злокачественным новообразованием. Заболевание развивается постепенно. До 5 лет врожденный дискератоз протекает латентно. Первые проявления могут развиваться в возрасте от 6 до 15 лет, иногда - после 20. С возрастом происходит усугубление всех симптомов и развитие осложнений.
Первичным проявлением врожденного дискератоза, как правило, выступает дистрофия ногтевых пластинок. Ногти становятся тонкими и ломкими, возникает продольная исчерченность. Спустя 2-3 года к данным проявлениям присоединяется сетчатая гиперпигментация кожи с характерной локализацией - лицо, шея, верхняя часть поверхности грудной клетки. Средний размер пораженных участков - 2-8 мм в диаметре. Кожа атрофируется, приобретает коричневый оттенок, на ней могут наблюдаться телеангиэктазии. Часто возникает гипергидроз и гиперкератоз стоп и ладоней, акроцианоз, алопеция. Спустя некоторое время ногтевые пластики могут самопроизвольно отпадать, что чаще всего наблюдается на мизинцах стоп.
Ассоциированные симптомы: Лейкоцитоз. Нейтропения. Потливость головы.
Первичным проявлением врожденного дискератоза, как правило, выступает дистрофия ногтевых пластинок. Ногти становятся тонкими и ломкими, возникает продольная исчерченность. Спустя 2-3 года к данным проявлениям присоединяется сетчатая гиперпигментация кожи с характерной локализацией - лицо, шея, верхняя часть поверхности грудной клетки. Средний размер пораженных участков - 2-8 мм в диаметре. Кожа атрофируется, приобретает коричневый оттенок, на ней могут наблюдаться телеангиэктазии. Часто возникает гипергидроз и гиперкератоз стоп и ладоней, акроцианоз, алопеция. Спустя некоторое время ногтевые пластики могут самопроизвольно отпадать, что чаще всего наблюдается на мизинцах стоп.
Ассоциированные симптомы: Лейкоцитоз. Нейтропения. Потливость головы.
Диагностика
Диагностика врожденного дискератоза затруднена. Заболевание встречается крайне редко и имеет большое количество клинических проявлений. Диагноз выставляется на основании всех имеющихся симптомов и синдромов, данных лабораторных анализов и гистологического исследования биоптата кожных покровов.
Общие клинические анализы позволяют установить наличие отдельных синдромов. В ОАК на ранних стадиях возможен лейкоцитоз, нейтропения, повышение СОЭ. Далее развивается панцитопения, апластическая анемия, возможны макроцитоз и повышение концентрации фетального гемоглобина. Стернальная пункция на поздних стадиях выявляет депрессию костного мозга. Гистологическое исследование пораженных участков кожи позволяет обнаружить атрофию эпидермиса, слабовыраженный гиперкератоз, увеличение пигментации базального слоя, в дерме - увеличение количества меланофагов в сосочковом и сетчатом слоях, расширение сосудов микроциркуляторного русла. Возможно нарушение структуры коллагена, гомогенизация и фрагментация его волокон. Специфическая диагностика направлена на выявление дефектов кариотипа. Проводится кариотипирование или флуоресцентная гибридизация, при которых удается установить мутацию гена в участке Xq28, редко - в 3q14.
Общие клинические анализы позволяют установить наличие отдельных синдромов. В ОАК на ранних стадиях возможен лейкоцитоз, нейтропения, повышение СОЭ. Далее развивается панцитопения, апластическая анемия, возможны макроцитоз и повышение концентрации фетального гемоглобина. Стернальная пункция на поздних стадиях выявляет депрессию костного мозга. Гистологическое исследование пораженных участков кожи позволяет обнаружить атрофию эпидермиса, слабовыраженный гиперкератоз, увеличение пигментации базального слоя, в дерме - увеличение количества меланофагов в сосочковом и сетчатом слоях, расширение сосудов микроциркуляторного русла. Возможно нарушение структуры коллагена, гомогенизация и фрагментация его волокон. Специфическая диагностика направлена на выявление дефектов кариотипа. Проводится кариотипирование или флуоресцентная гибридизация, при которых удается установить мутацию гена в участке Xq28, редко - в 3q14.
Дифференциальная диагностика
Дифференциальная диагностика врожденного дискератоза проводится с пигментной ксеродермой, ангидротической эктодермальной дисплазией, пойкилодермией, буллезным эпидермолизом, анемией Фанкони.
Лечение
Лечение врожденного дискератоза малоэффективно. Основная цель - борьба с угнетением функции костного мозга. Метод выбора - трансплантация костного мозга от родного брата или сестры, не страдающих данным заболеванием. Даже на фоне пересадки у большей части больных наблюдается реакция «трансплантат против хозяина». При невозможности провести трансплантацию показано использование анаболических гормональных средств (оксиметолон), колониестимулирующих факторов, эритропоэтинов. Лечение злокачественных новообразований у таких больных проводится при помощи протонной терапии. При лейкоплакии используются ретиноиды и Р-каротин.
Прогноз при врожденном дискератозе неблагоприятный. Почти всегда заболевание имеет летальный исход. К смерти приводят злокачественные новообразования, присоединение вторичной инфекции (в основном - пневмонии), желудочно-кишечные или другие внутренние кровотечения. Чаще всего пациенты умирают от плоскоклеточного рака ротовой полости, гортаноглотки, половых органов, мочеиспускательного канала и ЖКТ, реже - аденокарциномы поджелудочной железы, лимфомы Ходжкина.
Прогноз при врожденном дискератозе неблагоприятный. Почти всегда заболевание имеет летальный исход. К смерти приводят злокачественные новообразования, присоединение вторичной инфекции (в основном - пневмонии), желудочно-кишечные или другие внутренние кровотечения. Чаще всего пациенты умирают от плоскоклеточного рака ротовой полости, гортаноглотки, половых органов, мочеиспускательного канала и ЖКТ, реже - аденокарциномы поджелудочной железы, лимфомы Ходжкина.
|
|