Pharmacological Group
Latin name
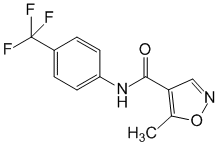
Leflunomidum ( Leflunomidi).
Chemical name
5-Метил-N-[4-(трифторметил)фенил]-4-изоксазолкарбоксамид.
Used in the treatment
CAS Code
75706-12-6.
Pharmacological action
Фармакологическое действие -.
Антипролиферативное, иммунодепрессивное, противовоспалительное, противоревматическое.
Антипролиферативное, иммунодепрессивное, противовоспалительное, противоревматическое.
Characteristics of the substance
Ингибитор синтеза пиримидинов. Молекулярная масса - 270,2.
Pharmacodynamics
Механизм действия обусловлен ингибированием дигидрооротатдегидрогеназы - фермента, участвующего в синтезе пиримидинов.
Pharmacokinetics
После приема внутрь лефлуномид метаболизируется в активный метаболит А77 1726 (далее обозначаемый как М1), который обуславливает действие препарата in vivo.
Лефлуномид в плазме крови выявляется в следовых количествах, поэтому при фармакокинетических исследованиях определялись концентрации М1.
После приема внутрь сmax достигается через 6-12 Время полужизни в плазме длительное (Т1/2 около 2 нед). Для быстрого достижения равновесной концентрации М1 в плазме крови в клинических испытаниях использовалось применение нагрузочной дозы препарата по 100 мг в течение 3 дней. Показано, что без предварительной нагрузочной дозы концентрация М1 в плазме достигает равновесных значений в течение 2 месяцев. Результирующие концентрации М1 в плазме крови, определяемые после приема нагрузочных и последующих терапевтических доз лефлуномида, показывают, что плазматические уровни М1 являются дозозависимыми. Определение фармакокинетических параметров М1 проводили после приема внутрь лефлуномида в суточных дозах 5, 10 и 25 мг в день (предварительные нагрузочные дозы - 50, 100 и 100 мг соответственно) у больных ревматоидным артритом (n=54). Через 24 ч после нагрузочной дозы концентрации М1 составляли 4,0±0,6; 8,4±2,1 и 8,5±2,2 мкг/мл соответственно; через 24 ч после приема терапевтических доз и в состоянии равновесия - 8,8±2,9; 18±9,6 и 63±36 мкг/мл.
Биодоступность таблетированной формы лефлуномида - 80%. Совместный прием внутрь таблеток лефлуномида и жирной пищи не оказывает существенного влияния на уровень М1 в плазме крови.
М1 имеет низкий объем распределения (0,13л/кг) и высокую степень связывания с плазматическим альбумином (>99,3%) у здоровых пациентов. Показано, что связывание с белками является линейным при терапевтических концентрациях. Уровень несвязанной фракции М1 несколько выше у больных ревматоидным артритом и приблизительно удваивается у пациентов с хронической почечной недостаточностью. Механизм и значение этих повышений неизвестны.
Лефлуномид метаболизируется до одного главного (М1) и нескольких второстепенных метаболитов. Из второстепенных метаболитов только 4-трифторметилаланин (ТФМА) количественно определяется у некоторых пациентов в низких концентрациях. Родственные соединения редко определяются в плазме. В настоящее время не выявлены специфические места метаболизма лефлуномида. На основании исследований in vivo и in vitro можно предположить участие в этом процессе гастроинтестинальной стенки и печени. Специфические ферменты метаболизма лефлуномида не выделены, однако показано участие цитоплазматической и микросомальной фракций клеток печени в метаболизме соединения.
Активный метаболит М1 выводится как путем последующего метаболизма и экскреции почками, так и посредством прямого выведения с желчью. В 28-дневном исследовании путей выведения соединения (n=3), с использованием однократной дозы радиоактивно меченного препарата, показано, что около 43% радиоактивной дозы выводится с мочой и 48% - с фекалиями. Последующий анализ образцов показал, что основными метаболитами в моче являются глюкурониды лефлуномида и дериваты М1. Основным метаболитом в фекалиях являлся М1. Из этих двух путей выведение с мочой было более значимым в первые 96 после чего доминировал второй путь экскреции. В исследовании с в/в введением М1 клиренс был равен 31 мл/.
В малых исследованиях с использованием активированного угля (n=1) и колестирамина (n=3) для облегчения выведения ЛС значения Т1/2 М1 из плазмы in vivo снижались с более чем 1 недели до приблизительно 1 дня. Сходные снижения Т1/2 наблюдались у группы добровольцев (n=96), участвовавших в исследованиях с колестираминовой нагрузкой. Этот факт дает основания предполагать, что повторное всасывание из желчи вносит основной вклад в поддержание длительного Т1/2 М1 из плазмы. Исследования, проводимые при применении гемодиализа и амбулаторного перитонеального диализа, показывают, что М1 не диализируется из плазмы и перитонеальной жидкости.
Зависимость параметров фармакокинетики от некоторых факторов.
Достоверных половых и возрастных различий в фармакокинетике М1 in vivo не выявлено.
Популяционный фармакокинетический анализ в Фазе III исследований показывает, что у курильщиков клиренс возрастает на 38% по сравнению с некурящими; однако нет различий в клинической эффективности (лефлуномида) у курящих и некурящих.
У больных с хронической почечной недостаточностью, нуждающихся в гемодиализе или лабораторном перитонеальном диализе (n=6), после однократного приема ЛС не выявлено значимого влияния на уровни циркулирующего М1. Фракция несвязанного М1 была практически удвоена, но механизм этого возрастания не известен. В свете того, что почки участвуют в процессе выведения препарата и при отсутствии адекватных исследований при применении лефлуномида у больных с почечной недостаточностью, следует соблюдать осторожность при назначении лечения этим препаратом.
Исследования влияния печеночной недостаточности на фармакокинетику М1 не были проведены. Принимая во внимание метаболизм лефлуномида в активные метаболиты, роль печени в элиминации и рециркуляции в организме лекарств, возможный риск возрастания гепатотоксичности, применение лефлуномида у больных с печеночной недостаточностью не рекомендуется.
Канцерогенность, мутагенность и влияние на фертильность.
В двухлетних исследованиях на крысах канцерогенность лефлуномида не была выявлена при пероральных дозах, вплоть до максимально переносимой - 6 мг/кг (приблизительно 1/40 максимальной системной экспозиции М1 у человека, основанной на AUC). Однако в двухгодичных исследованиях на мышах-самцах наблюдалось увеличение случаев образования лимфомы при максимальной исследованной пероральной дозировке 15 мг/кг (1,7-кратной экспозиции М1 у человека, основанной на AUC). В тех же исследованиях у мышей-самок наблюдалось сочетанное, дозозависимое повышение образования бронхоальвеолярных аденом и карцином при возрастающих дозах, начиная с 1,5 мг/кг (приблизительно 1/10 экспозиции М1 у человека, основанной на AUC). Значение этих данных относительно клинического использования лефлуномида не определено.
Лефлуномид не проявлял мутагенности в тесте Эймса, тесте внепланового синтеза ДНК и/или тесте с гипоксантин-гуанинфосфорибозилтрансферазой клеток яичника китайского хомячка. Также лефлуномид не проявлял кластогенных свойств в микроядерном тесте на мышах in vivo и в цитогенетическом тесте на клетках костного мозга китайских хомячков in vivo. Однако 4-трифторметиланилин (ТФМА), второстепенный метаболит лефлуномида, проявлял мутагенные свойства в тесте Эймса и в тесте с гипоксантин-гуанинфосфорибозилтрансферазой клеток яичника китайского хомячка и показал кластогенные свойства в тесте хромосомных аберраций на клетках китайского хомячка in vitro. ТФМА не показал кластогенных свойств в микроядерном тесте на мышах in vivo или цитогенетическом тесте на клетках костного мозга китайского хомячка in vivo.
Лефлуномид не влияет на фертильность самцов и самок крыс при пероральных дозировках до 4,0 мг/кг (ориентировочно 1/30 экспозиции М1 у человека, основанной на AUC).
Клинические испытания.
Эффективность лефлуномида в лечении ревматоидного артрита (РА) была продемонстрирована в 3 контролируемых испытаниях. Облегчение симптоматики оценивалось с помощью Критерия ответа ACR20, который является комплексом клинических, лабораторных и функциональных исследований при РА (ACR20 Responder Index, ACR - American сollege of Rheumatology - Американская коллегия ревматологов). «ACR20 респондером» является пациент с РА, у которого наблюдается 20% или более улучшение при оценке числа болезненных/воспаленных суставов и в 3 из следующих 5 критериев: общей оценки состояния врачом, общей оценки состояния пациентом, показателя инвалидизации (по модифицированной анкете оценки здоровья - MHAQ- Modified Health Assessment Questionnaire), визуального аналога болевой шкалы и показателя СОЭ или концентрации С-реактивного белка. «ACR20 респондером в конечной точке» является пациент, у которого на момент окончания исследований наблюдалось 20% или более улучшение по указанным выше параметрам. Задержка в прогрессировании структурных повреждений суставов в сравнении с контролем определялась с помощью шкалы Шарпа путем общего подсчета краевых эрозий и сужений суставных щелей кистей/запястий и переднего отдела стоп.
В начале клинических испытаний все пациенты, которым был назначен лефлуномид, принимали нагрузочную дозу 100 мг/день в течение 3 дней.
В клинических испытаниях US301 в США были рандомизированы 482 пациента с активной формой РА продолжительностью не менее 6 мес. Пациенты получали лефлуномид в дозировке 20 мг/день (n=182), или метотрексат с возрастающей дозировкой от 7,5 до 15 мг в неделю (n=182), или плацебо (n=118). Все пациенты принимали фолиевую кислоту по 1 мг дважды в день. Время лечения составляло 52 нед.
В исследованиях MN301 в Европе были рандомизированы 358 пациентов с активной формой РА, получавшие или лефлуномид 20 мг/день (n=133), или сульфасалазин 2,0 г/день (n=133), или плацебо (n=92). Продолжительность лечения - 24 нед. Клинические испытания MN303 в Европе являлись дополнительным 6-месячным плацебо-неконтролируемым продолжением исследований MN301, результатом которых было сравнение эффективности лефлуномида и сульфасалазина в течение 12 мес.
В клинических испытаниях MN302 в Европе были рандомизированы 999 пациентов с активной формой РА, получавшие или лефлуномид по 20 мг/день (n=501), или метотрексат в возрастающей дозировке от 7,5 до 15 мг в неделю (n=498). 10% пациентов дополнительно принимали фолиевую кислоту. Время лечения составляло 52 нед.
Результаты клинических испытаний.
По данным Критерия ответа ACR20 в конечной точке, количество больных РА с улучшениями при лечении лефлуномидом было статистически достоверно выше в сравнении с ответом на плацебо: 41% и 18% соответственно в группе US301, 49% и 28% соответственно в группах MN301/303. По этой модификации Критерия ответа ACR20 эффективность лечения была стабильной с течением времени и составляла через 6 мес 41%, а через 12 мес - 49%. Достоверных различий в эффективности лечения между лефлуномидом и метотрексатом и между лефлуномидом и сульфасалазином не выявлено.
По основной модификации ACR20 статистически значимый эффект лечения лефлуномидом у группы US301 в сравнении с плацебо определялся уже через 1 мес после начала терапии (38 и 20% соответственно), достигал максимума и стабилизировался на 3-6 мес (50-55 и 26-30% соответственно) и практически не изменялся в течение последующих 6 мес.
В таблице 1 представлены в сравнении количества (в % от общего числа) ACR20, ACR50 и ACR70 отвечающих пациентов с РА, у которых наблюдалось, соответственно, 20, 50 или 70% интегральное улучшение по исследуемым параметрам (табл. 2).
Таблица 1.
Сравнительные результаты клинических исследований лефлуномида.
1 - p<0,001 (лефлуномид к плацебо).
2 - p<0,02 (лефлуномид к плацебо).
Таблица 2.
Средние изменения отдельных параметров исследований, включенных в Критерий ответа ACR.
1 - подсчет состояния 28 суставов.
2 - визуальный аналог болевой шкалы (0 - нет боли, 10 - максимум).
По данным рентгенологических исследований структурных поражений суставов, оцениваемых по шкале Шарпа, лечение лефлуномидом по сравнению с плацебо статистически достоверно замедляет скорость прогрессирования РА. У пациентов группы US301 этот параметр составил 0,5 единицы в год при скорости 2,2 единицы в случае плацебо, а в исследованиях MN301/303 наблюдалась стабилизация процесса, тогда как при приеме плацебо скорость рентгенологического прогрессирования была 5,5 единиц в год. Достоверных различий при лечении лефлуномидом и метотрексатом или лефлуномидом и сульфасалазином не наблюдалось.
Лефлуномид в плазме крови выявляется в следовых количествах, поэтому при фармакокинетических исследованиях определялись концентрации М1.
После приема внутрь сmax достигается через 6-12 Время полужизни в плазме длительное (Т1/2 около 2 нед). Для быстрого достижения равновесной концентрации М1 в плазме крови в клинических испытаниях использовалось применение нагрузочной дозы препарата по 100 мг в течение 3 дней. Показано, что без предварительной нагрузочной дозы концентрация М1 в плазме достигает равновесных значений в течение 2 месяцев. Результирующие концентрации М1 в плазме крови, определяемые после приема нагрузочных и последующих терапевтических доз лефлуномида, показывают, что плазматические уровни М1 являются дозозависимыми. Определение фармакокинетических параметров М1 проводили после приема внутрь лефлуномида в суточных дозах 5, 10 и 25 мг в день (предварительные нагрузочные дозы - 50, 100 и 100 мг соответственно) у больных ревматоидным артритом (n=54). Через 24 ч после нагрузочной дозы концентрации М1 составляли 4,0±0,6; 8,4±2,1 и 8,5±2,2 мкг/мл соответственно; через 24 ч после приема терапевтических доз и в состоянии равновесия - 8,8±2,9; 18±9,6 и 63±36 мкг/мл.
Биодоступность таблетированной формы лефлуномида - 80%. Совместный прием внутрь таблеток лефлуномида и жирной пищи не оказывает существенного влияния на уровень М1 в плазме крови.
М1 имеет низкий объем распределения (0,13л/кг) и высокую степень связывания с плазматическим альбумином (>99,3%) у здоровых пациентов. Показано, что связывание с белками является линейным при терапевтических концентрациях. Уровень несвязанной фракции М1 несколько выше у больных ревматоидным артритом и приблизительно удваивается у пациентов с хронической почечной недостаточностью. Механизм и значение этих повышений неизвестны.
Лефлуномид метаболизируется до одного главного (М1) и нескольких второстепенных метаболитов. Из второстепенных метаболитов только 4-трифторметилаланин (ТФМА) количественно определяется у некоторых пациентов в низких концентрациях. Родственные соединения редко определяются в плазме. В настоящее время не выявлены специфические места метаболизма лефлуномида. На основании исследований in vivo и in vitro можно предположить участие в этом процессе гастроинтестинальной стенки и печени. Специфические ферменты метаболизма лефлуномида не выделены, однако показано участие цитоплазматической и микросомальной фракций клеток печени в метаболизме соединения.
Активный метаболит М1 выводится как путем последующего метаболизма и экскреции почками, так и посредством прямого выведения с желчью. В 28-дневном исследовании путей выведения соединения (n=3), с использованием однократной дозы радиоактивно меченного препарата, показано, что около 43% радиоактивной дозы выводится с мочой и 48% - с фекалиями. Последующий анализ образцов показал, что основными метаболитами в моче являются глюкурониды лефлуномида и дериваты М1. Основным метаболитом в фекалиях являлся М1. Из этих двух путей выведение с мочой было более значимым в первые 96 после чего доминировал второй путь экскреции. В исследовании с в/в введением М1 клиренс был равен 31 мл/.
В малых исследованиях с использованием активированного угля (n=1) и колестирамина (n=3) для облегчения выведения ЛС значения Т1/2 М1 из плазмы in vivo снижались с более чем 1 недели до приблизительно 1 дня. Сходные снижения Т1/2 наблюдались у группы добровольцев (n=96), участвовавших в исследованиях с колестираминовой нагрузкой. Этот факт дает основания предполагать, что повторное всасывание из желчи вносит основной вклад в поддержание длительного Т1/2 М1 из плазмы. Исследования, проводимые при применении гемодиализа и амбулаторного перитонеального диализа, показывают, что М1 не диализируется из плазмы и перитонеальной жидкости.
Зависимость параметров фармакокинетики от некоторых факторов.
Достоверных половых и возрастных различий в фармакокинетике М1 in vivo не выявлено.
Популяционный фармакокинетический анализ в Фазе III исследований показывает, что у курильщиков клиренс возрастает на 38% по сравнению с некурящими; однако нет различий в клинической эффективности (лефлуномида) у курящих и некурящих.
У больных с хронической почечной недостаточностью, нуждающихся в гемодиализе или лабораторном перитонеальном диализе (n=6), после однократного приема ЛС не выявлено значимого влияния на уровни циркулирующего М1. Фракция несвязанного М1 была практически удвоена, но механизм этого возрастания не известен. В свете того, что почки участвуют в процессе выведения препарата и при отсутствии адекватных исследований при применении лефлуномида у больных с почечной недостаточностью, следует соблюдать осторожность при назначении лечения этим препаратом.
Исследования влияния печеночной недостаточности на фармакокинетику М1 не были проведены. Принимая во внимание метаболизм лефлуномида в активные метаболиты, роль печени в элиминации и рециркуляции в организме лекарств, возможный риск возрастания гепатотоксичности, применение лефлуномида у больных с печеночной недостаточностью не рекомендуется.
Канцерогенность, мутагенность и влияние на фертильность.
В двухлетних исследованиях на крысах канцерогенность лефлуномида не была выявлена при пероральных дозах, вплоть до максимально переносимой - 6 мг/кг (приблизительно 1/40 максимальной системной экспозиции М1 у человека, основанной на AUC). Однако в двухгодичных исследованиях на мышах-самцах наблюдалось увеличение случаев образования лимфомы при максимальной исследованной пероральной дозировке 15 мг/кг (1,7-кратной экспозиции М1 у человека, основанной на AUC). В тех же исследованиях у мышей-самок наблюдалось сочетанное, дозозависимое повышение образования бронхоальвеолярных аденом и карцином при возрастающих дозах, начиная с 1,5 мг/кг (приблизительно 1/10 экспозиции М1 у человека, основанной на AUC). Значение этих данных относительно клинического использования лефлуномида не определено.
Лефлуномид не проявлял мутагенности в тесте Эймса, тесте внепланового синтеза ДНК и/или тесте с гипоксантин-гуанинфосфорибозилтрансферазой клеток яичника китайского хомячка. Также лефлуномид не проявлял кластогенных свойств в микроядерном тесте на мышах in vivo и в цитогенетическом тесте на клетках костного мозга китайских хомячков in vivo. Однако 4-трифторметиланилин (ТФМА), второстепенный метаболит лефлуномида, проявлял мутагенные свойства в тесте Эймса и в тесте с гипоксантин-гуанинфосфорибозилтрансферазой клеток яичника китайского хомячка и показал кластогенные свойства в тесте хромосомных аберраций на клетках китайского хомячка in vitro. ТФМА не показал кластогенных свойств в микроядерном тесте на мышах in vivo или цитогенетическом тесте на клетках костного мозга китайского хомячка in vivo.
Лефлуномид не влияет на фертильность самцов и самок крыс при пероральных дозировках до 4,0 мг/кг (ориентировочно 1/30 экспозиции М1 у человека, основанной на AUC).
Клинические испытания.
Эффективность лефлуномида в лечении ревматоидного артрита (РА) была продемонстрирована в 3 контролируемых испытаниях. Облегчение симптоматики оценивалось с помощью Критерия ответа ACR20, который является комплексом клинических, лабораторных и функциональных исследований при РА (ACR20 Responder Index, ACR - American сollege of Rheumatology - Американская коллегия ревматологов). «ACR20 респондером» является пациент с РА, у которого наблюдается 20% или более улучшение при оценке числа болезненных/воспаленных суставов и в 3 из следующих 5 критериев: общей оценки состояния врачом, общей оценки состояния пациентом, показателя инвалидизации (по модифицированной анкете оценки здоровья - MHAQ- Modified Health Assessment Questionnaire), визуального аналога болевой шкалы и показателя СОЭ или концентрации С-реактивного белка. «ACR20 респондером в конечной точке» является пациент, у которого на момент окончания исследований наблюдалось 20% или более улучшение по указанным выше параметрам. Задержка в прогрессировании структурных повреждений суставов в сравнении с контролем определялась с помощью шкалы Шарпа путем общего подсчета краевых эрозий и сужений суставных щелей кистей/запястий и переднего отдела стоп.
В начале клинических испытаний все пациенты, которым был назначен лефлуномид, принимали нагрузочную дозу 100 мг/день в течение 3 дней.
В клинических испытаниях US301 в США были рандомизированы 482 пациента с активной формой РА продолжительностью не менее 6 мес. Пациенты получали лефлуномид в дозировке 20 мг/день (n=182), или метотрексат с возрастающей дозировкой от 7,5 до 15 мг в неделю (n=182), или плацебо (n=118). Все пациенты принимали фолиевую кислоту по 1 мг дважды в день. Время лечения составляло 52 нед.
В исследованиях MN301 в Европе были рандомизированы 358 пациентов с активной формой РА, получавшие или лефлуномид 20 мг/день (n=133), или сульфасалазин 2,0 г/день (n=133), или плацебо (n=92). Продолжительность лечения - 24 нед. Клинические испытания MN303 в Европе являлись дополнительным 6-месячным плацебо-неконтролируемым продолжением исследований MN301, результатом которых было сравнение эффективности лефлуномида и сульфасалазина в течение 12 мес.
В клинических испытаниях MN302 в Европе были рандомизированы 999 пациентов с активной формой РА, получавшие или лефлуномид по 20 мг/день (n=501), или метотрексат в возрастающей дозировке от 7,5 до 15 мг в неделю (n=498). 10% пациентов дополнительно принимали фолиевую кислоту. Время лечения составляло 52 нед.
Результаты клинических испытаний.
По данным Критерия ответа ACR20 в конечной точке, количество больных РА с улучшениями при лечении лефлуномидом было статистически достоверно выше в сравнении с ответом на плацебо: 41% и 18% соответственно в группе US301, 49% и 28% соответственно в группах MN301/303. По этой модификации Критерия ответа ACR20 эффективность лечения была стабильной с течением времени и составляла через 6 мес 41%, а через 12 мес - 49%. Достоверных различий в эффективности лечения между лефлуномидом и метотрексатом и между лефлуномидом и сульфасалазином не выявлено.
По основной модификации ACR20 статистически значимый эффект лечения лефлуномидом у группы US301 в сравнении с плацебо определялся уже через 1 мес после начала терапии (38 и 20% соответственно), достигал максимума и стабилизировался на 3-6 мес (50-55 и 26-30% соответственно) и практически не изменялся в течение последующих 6 мес.
В таблице 1 представлены в сравнении количества (в % от общего числа) ACR20, ACR50 и ACR70 отвечающих пациентов с РА, у которых наблюдалось, соответственно, 20, 50 или 70% интегральное улучшение по исследуемым параметрам (табл. 2).
Таблица 1.
Сравнительные результаты клинических исследований лефлуномида.
| Исследуемая группа | Критерий ответа | ||
| ACR 20% | ACR 50% | ACR 70% | |
| Плацебо-контролируемые исследования | |||
| US301 (12 мес) | |||
| лефлуномид (n=178) | 52,21 | 34,31 | 20,21 |
| плацебо (n=118) | 26,3 | 7,6 | 4,2 |
| метотрексат (n=180) | 45,6 | 22,8 | 9,4 |
| MN301 (6 мес) | |||
| лефлуномид (n=130) | 54,62 | 33,12 | 10,02 |
| плацебо (n=91) | 28,6 | 14,3 | 2,2 |
| сульфасалазин (n=132) | 56,8 | 30,3 | 7,6 |
| Плацебо-неконтролируемые исследования | |||
| MN302 (12 мес) | |||
| лефлуномид (n=495) | 51,1 | 31,1 | 9,9 |
| метотрексат (n=489) | 65,2 | 43,8 | 16,4 |
1 - p<0,001 (лефлуномид к плацебо).
2 - p<0,02 (лефлуномид к плацебо).
Таблица 2.
Средние изменения отдельных параметров исследований, включенных в Критерий ответа ACR.
| Параметры | Плацебо-контролируемые исследования | Плацебо-неконтролируемые исследования | ||||||
| US301 | MN301 | MN302 | ||||||
| Лефлуномид | Метотрексат | Плацебо | Лефлуномид | Метотрексат | Плацебо | Лефлуномид | Метотрексат | |
| Число болезненных суставов1 | -7,7 | -6,6 | -3,0 | -9,7 | -8,1 | -4,3 | -8,3 | -9,7 |
| Число воспаленных суставов1 | -5,7 | -5,4 | -2,9 | -7,2 | -6,2 | -3,4 | -6,8 | -9,0 |
| Оценка пациентом | -2,1 | -1,5 | 0,1 | -2,8 | -2,6 | -0,9 | -2,3 | -3,0 |
| Оценка врачом | -2,8 | -2,4 | -1,0 | -2,7 | -2,5 | -0,8 | -2,3 | -3,1 |
| Инвалидизация | -0,29 | -0,15 | 0,07 | -0,50 | -0,29 | -0,04 | -0,37 | -0,44 |
| Интенсивность боли2 | -2,2 | -1,7 | -0,5 | -2,7 | -2,0 | -0,9 | -2,1 | -2,9 |
| СОЭ | -6,26 | -6,48 | 2,56 | -7,48 | -16,56 | 3,44 | -10,12 | -22,18 |
| С-реактивный белок | -0,62 | -0,50 | 0,47 | -2,26 | -1,19 | 0,16 | -1,86 | -2,45 |
1 - подсчет состояния 28 суставов.
2 - визуальный аналог болевой шкалы (0 - нет боли, 10 - максимум).
По данным рентгенологических исследований структурных поражений суставов, оцениваемых по шкале Шарпа, лечение лефлуномидом по сравнению с плацебо статистически достоверно замедляет скорость прогрессирования РА. У пациентов группы US301 этот параметр составил 0,5 единицы в год при скорости 2,2 единицы в случае плацебо, а в исследованиях MN301/303 наблюдалась стабилизация процесса, тогда как при приеме плацебо скорость рентгенологического прогрессирования была 5,5 единиц в год. Достоверных различий при лечении лефлуномидом и метотрексатом или лефлуномидом и сульфасалазином не наблюдалось.
Indications for use
По данным Physician Desk Reference (2005). Лефлуномид показан у взрослых для лечения активного РА. Снижения объективных симптомов и улучшения субъективного состояния больных. Задержки образования краевых эрозий суставов и сужений суставных щелей. Выявляемых рентгеноскопически. Лечение ацетилсалициловой кислотой, НПВС и/или низкими дозами кортикостероидов может быть продолжено при терапии лефлуномидом. Комбинированное использование лефлуномида с противомалярийными препаратами. Препаратами золота для внутримышечного введения или приема внутрь. Пеницилламином. Азатиоприном и метотрексатом достаточно не исследовано.
Contraindications
Гиперчувствительность, беременность.
Restrictions on use
Возможность иммунодепрессии.
Лефлуномид не рекомендуется пациентам с тяжелыми иммунодефицитами, дисплазией костного мозга, тяжелыми, неконтролируемыми инфекциями.
В немногочисленных сообщениях указывалось на выявление панцитопении при приеме лефлуномида. В большинстве этих случаев больные получали совместное лечение метотрексатом или другими иммунодепрессивными препаратами, или терапия этими соединениями была отменена перед исследованием; в некоторых случаях у пациентов в анамнезе были выявлены существенные гематологические нарушения. При лечении таких пациентов лефлуномидом терапия должна проводиться с осторожностью и под постоянным клиническим и гематологическим контролем. Использование лефлуномида в комбинированной терапии с метотрексатом в контролируемых исследованиях адекватно не изучено.
При выявлении угнетения костного мозга у пациента, принимающего лефлуномид, лечение этим препаратом должно быть прекращено, и для быстрого снижения в плазме крови концентрации активного метаболита лефлуномида необходимо использовать колестирамин или активированный уголь ( см «Фармакология. Фармакокинетика», «Меры предосторожности. Необходимость в выведении препарата»).
В случае, когда принято решение замены лефлуномида другим противоревматическим препаратом с известными гематодепрессивными свойствами, является разумным продолжать контролировать гематотоксичность из-за возможного взаимного наложения системного действия обоих препаратов. Выведение лефлуномида с помощью колестирамина или активированного угля может снизить этот риск, но может и ухудшить состояние больного, восприимчивого к лечению лефлуномидом.
Кожные реакции.
Имеются сообщения об отдельных случаях возникновения синдрома Стивенса - Джонсона и токсического эпидермального некролиза у больных, принимавших лефлуномид. В случае развития у пациента, принимающего лефлуномид, одного из этих состояний, необходимо прервать терапию лефлуномидом и провести рекомендованную процедуру выведения препарата ( см «Фармакология. Фармакокинетика», «Меры предосторожности. Необходимость в выведении препарата»).
Гепатотоксичность.
В клинических испытаниях лечение лефлуномидом у определенного числа пациентов было связано с повышением активности печеночных ферментов, в первую очередь АЛТ и АСТ; эти эффекты в большинстве случаев были обратимыми. Большинство повышений активности трансаминаз было средним (?2-кратной верхней границы нормы, ВГН) и обычно снижалось при продолжении лечения. Существенные повышения (>3-кратной ВГН) выявлялись нечасто и были обратимыми при снижении дозировки или прерывании лечения. В таблице 3 представлены данные о повышении активности печеночных ферментов, определявшиеся при клинических испытаниях ежемесячно. Необходимо отметить, что отсутствие применения фолиевой кислоты в клинических испытаниях MN302 ассоциируется с большей частотой повышения активности печеночных ферментов при лечении метотрексатом.
Таблица 3.
Активность печеночных трансаминаз в клинических исследованиях.
*-только 10% пациентов в MN302 принимали фолат. Все пациенты в US301 получали фолат.
Уровень активности АЛТ должен быть основным диагностическим показателем наличия гепатотоксичности и в начале контролироваться с месячными интервалами, затем, при стабилизации, интервалы определяются по индивидуальной клинической ситуации. Рекомендуется следующее руководство регулирования дозировки или прерывания терапии, основывающееся на опасности и продолжительности подъема значения активности АЛТ: при постоянном повышении активности АЛТ >2-кратной ВГН, снижение дозы до 10 мг/день может позволить продолжить прием лефлуномида. Если, несмотря на снижение дозировки, подъем активности АЛТ продолжает сохраняться в границах >2-кратной, но ?3-кратной ВГН при желательности продолжения лечения рекомендуется проведение биопсии печени. Если повышение >3-кратной ВГН длительно сохраняется, несмотря на снижение дозировки, терапию лефлуномидом следует прервать, назначить прием колестирамина и, при необходимости, повторить прием колестирамина ( см «Фармакология. Фармакокинетика», «Меры предосторожности. Необходимость в выведении препарата»).
Наблюдались отдельные случаи повышения активности щелочной фосфатазы и концентрации билирубина в сыворотке крови.
Доклиническая стадия заболевания печени.
В связи с фактом вероятного риска усиления токсического действия на печень, ролью печени в активации, выведении и рециркуляции лекарств, применение лефлуномида не рекомендуется у пациентов с существенным нарушением функций печени или инфицированных вирусами гепатита в или с.
Малигнизация.
Риск малигнизации, особенно лимфопролиферативных заболеваний, возрастает с применением некоторых ЛС с иммунодепрессивным действием. Лефлуномид обладает возможностью иммунодепрессивного действия. В клинических испытаниях с применением лефлуномида не наблюдалось увеличения частоты малигнизации или возникновения лимфопролиферативных заболеваний, однако для определения возможного риска проявления этих процессов необходимы более широкие и длительные исследования.
Методика выведения препарата.
Рекомендуется следующая методика выведения препарата, позволяющая достигать его неопределяемого в плазме уровня (менее чем 0,02 мг/л или 0,02 мкг/мл) после прекращения лечения лефлуномидом:
1) прием колестирамина по 8 г 3 раза в день в течение 11 дней (необязательно принимать колестирамин строго 11 дней подряд, если нет необходимости быстрого снижения уровня М1 в плазме);
2) контролировать меньшие чем 0,2 мг/л (0,2 мкг/мл) уровни М1 в плазме 2 независимыми тестами по меньшей мере 14 дней. Если плазменные уровни выше чем 0,02 мг/л, необходим дополнительный прием колестирамина.
Без применения методики выведения плазматический уровень М1 может не достигнуть значений менее 0,02 мг/л в течение 2 лет, в зависимости от индивидуальных вариаций клиренса.
Лефлуномид не рекомендуется пациентам с тяжелыми иммунодефицитами, дисплазией костного мозга, тяжелыми, неконтролируемыми инфекциями.
В немногочисленных сообщениях указывалось на выявление панцитопении при приеме лефлуномида. В большинстве этих случаев больные получали совместное лечение метотрексатом или другими иммунодепрессивными препаратами, или терапия этими соединениями была отменена перед исследованием; в некоторых случаях у пациентов в анамнезе были выявлены существенные гематологические нарушения. При лечении таких пациентов лефлуномидом терапия должна проводиться с осторожностью и под постоянным клиническим и гематологическим контролем. Использование лефлуномида в комбинированной терапии с метотрексатом в контролируемых исследованиях адекватно не изучено.
При выявлении угнетения костного мозга у пациента, принимающего лефлуномид, лечение этим препаратом должно быть прекращено, и для быстрого снижения в плазме крови концентрации активного метаболита лефлуномида необходимо использовать колестирамин или активированный уголь ( см «Фармакология. Фармакокинетика», «Меры предосторожности. Необходимость в выведении препарата»).
В случае, когда принято решение замены лефлуномида другим противоревматическим препаратом с известными гематодепрессивными свойствами, является разумным продолжать контролировать гематотоксичность из-за возможного взаимного наложения системного действия обоих препаратов. Выведение лефлуномида с помощью колестирамина или активированного угля может снизить этот риск, но может и ухудшить состояние больного, восприимчивого к лечению лефлуномидом.
Кожные реакции.
Имеются сообщения об отдельных случаях возникновения синдрома Стивенса - Джонсона и токсического эпидермального некролиза у больных, принимавших лефлуномид. В случае развития у пациента, принимающего лефлуномид, одного из этих состояний, необходимо прервать терапию лефлуномидом и провести рекомендованную процедуру выведения препарата ( см «Фармакология. Фармакокинетика», «Меры предосторожности. Необходимость в выведении препарата»).
Гепатотоксичность.
В клинических испытаниях лечение лефлуномидом у определенного числа пациентов было связано с повышением активности печеночных ферментов, в первую очередь АЛТ и АСТ; эти эффекты в большинстве случаев были обратимыми. Большинство повышений активности трансаминаз было средним (?2-кратной верхней границы нормы, ВГН) и обычно снижалось при продолжении лечения. Существенные повышения (>3-кратной ВГН) выявлялись нечасто и были обратимыми при снижении дозировки или прерывании лечения. В таблице 3 представлены данные о повышении активности печеночных ферментов, определявшиеся при клинических испытаниях ежемесячно. Необходимо отметить, что отсутствие применения фолиевой кислоты в клинических испытаниях MN302 ассоциируется с большей частотой повышения активности печеночных ферментов при лечении метотрексатом.
Таблица 3.
Активность печеночных трансаминаз в клинических исследованиях.
| Параметры | US301 | MN301 | MN302* | |||||
| Лефлуномид | Плацебо | Метотрексат | Лефлуномид | Плацебо | Сульфасалазин | Лефлуномид | Метотрексат | |
| АЛТ >3 ВГН (n, %) Возврат к ?2 ВГН: | 8 (4,4) 8 | 3 (2,5) 3 | 5 (2,7) 5 | 2 (1,5) 2 | 1 (1,1) 1 | 2 (1,5) 2 | 13 (2,6) 12 | 83 (16,7) 82 |
| Время подъема 0-3 мес 4-6 мес 7-9 мес 10-12 мес | 6 1 1 - | 1 1 1 - | 1 3 1 - | 2 - - - | 1 - - - | 2 - - - | 7 1 - 5 | 27 34 16 6 |
| АСТ >3 ВГН (n, %) Возврат к ?2 ВГН: | 4 (2,2) 4 | 2 (1,7) 2 | 1 (0,6) 1 | 2 (1,5) 2 | 0 - | 5 (3,8) 4 | 7 (1,4) 5 | 29 (5,8) 29 |
| Время подъема 0-3 мес 4-6 мес 7-9 мес 10-12 мес | 2 1 1 - | 1 1 - - | - 1 - - | 2 - - - | - - - - | 4 1 - - | 3 1 - 3 | 10 11 8 - |
*-только 10% пациентов в MN302 принимали фолат. Все пациенты в US301 получали фолат.
Уровень активности АЛТ должен быть основным диагностическим показателем наличия гепатотоксичности и в начале контролироваться с месячными интервалами, затем, при стабилизации, интервалы определяются по индивидуальной клинической ситуации. Рекомендуется следующее руководство регулирования дозировки или прерывания терапии, основывающееся на опасности и продолжительности подъема значения активности АЛТ: при постоянном повышении активности АЛТ >2-кратной ВГН, снижение дозы до 10 мг/день может позволить продолжить прием лефлуномида. Если, несмотря на снижение дозировки, подъем активности АЛТ продолжает сохраняться в границах >2-кратной, но ?3-кратной ВГН при желательности продолжения лечения рекомендуется проведение биопсии печени. Если повышение >3-кратной ВГН длительно сохраняется, несмотря на снижение дозировки, терапию лефлуномидом следует прервать, назначить прием колестирамина и, при необходимости, повторить прием колестирамина ( см «Фармакология. Фармакокинетика», «Меры предосторожности. Необходимость в выведении препарата»).
Наблюдались отдельные случаи повышения активности щелочной фосфатазы и концентрации билирубина в сыворотке крови.
Доклиническая стадия заболевания печени.
В связи с фактом вероятного риска усиления токсического действия на печень, ролью печени в активации, выведении и рециркуляции лекарств, применение лефлуномида не рекомендуется у пациентов с существенным нарушением функций печени или инфицированных вирусами гепатита в или с.
Малигнизация.
Риск малигнизации, особенно лимфопролиферативных заболеваний, возрастает с применением некоторых ЛС с иммунодепрессивным действием. Лефлуномид обладает возможностью иммунодепрессивного действия. В клинических испытаниях с применением лефлуномида не наблюдалось увеличения частоты малигнизации или возникновения лимфопролиферативных заболеваний, однако для определения возможного риска проявления этих процессов необходимы более широкие и длительные исследования.
Методика выведения препарата.
Рекомендуется следующая методика выведения препарата, позволяющая достигать его неопределяемого в плазме уровня (менее чем 0,02 мг/л или 0,02 мкг/мл) после прекращения лечения лефлуномидом:
1) прием колестирамина по 8 г 3 раза в день в течение 11 дней (необязательно принимать колестирамин строго 11 дней подряд, если нет необходимости быстрого снижения уровня М1 в плазме);
2) контролировать меньшие чем 0,2 мг/л (0,2 мкг/мл) уровни М1 в плазме 2 независимыми тестами по меньшей мере 14 дней. Если плазменные уровни выше чем 0,02 мг/л, необходим дополнительный прием колестирамина.
Без применения методики выведения плазматический уровень М1 может не достигнуть значений менее 0,02 мг/л в течение 2 лет, в зависимости от индивидуальных вариаций клиренса.
Use during pregnancy and lactation
Лефлуномид может нанести вред плоду при приеме беременными женщинами. Лефлуномид обладал тератогенностью при пероральном введении крысам во время органогенеза в дозе 15 мг/кг (в основном анофтальмия или микрофтальмия и внутренняя гидроцефалия). Системная экспозиция у крыс при этой дозе составляла приблизительно 1/10 уровня экспозиции, основанной на AUC, у человека. При этих условиях лефлуномид также вызывал снижение веса тела самок и усиление эмбриолетальности и снижение веса плодов выживших новорожденных. У кроликов пероральное введение лефлуномида в дозе 10 мг/кг во время органогенеза приводило к образованию сращенных диспластичных сегментов грудины. Уровень экспозиции при этой дозе был практически равен максимальному уровню экспозиции, основанной на AUC, у человека. При дозе 1 мг/кг лефлуномид не проявлял тератогенности у крыс и кроликов.
При воздействии на самок-крыс лефлуномидом в дозе 1,25 мг/кг, начиная за 14 дней до скрещивания и кончая завершением лактации, наблюдается снижение выживаемости потомства более чем на 90%. Уровень системной экспозиции при дозе 1,25 мг/кг был около 1/100 уровня экспозиции у человека, основанной на AUC.
Применение у женщин детородного возраста. Адекватных и строго контролированных исследований по оценке действия лефлуномида у беременных женщин проведено не было. Однако основываясь на данных исследований на животных, лефлуномид может повышать риск гибели плода при приеме в период беременности. Женщинам детородного возраста нельзя начинать лечение лефлуномидом до исключения беременности и (или) подтверждения использования надежной контрацепции. Перед началом лечения лефлуномидом пациентки должны быть в полном объеме проконсультированы о потенциальной серьезной опасности для плода.
Пациентки должны быть предупреждены, что при любой задержке наступления менструации или другой причине, предполагающей беременность, они должны немедленно известить врача для прохождения теста на беременность, и, при положительном результате, врач и пациентка должны обсудить риск для беременности. Вероятно, что быстрое снижение уровня активного метаболита в крови при использовании методики выведения препарата, при первой задержке менструации, сможет снизить риск применения лефлуномида для плода.
Всем женщинам детородного возраста после прекращения лечения лефлуномидом рекомендуется пройти процедуру выведения препарата. Женщинам, проходящим лечение лефлуномидом и желающим забеременеть, необходимо прервать прием препарата и пройти процедуру его выведения, включающую подтверждение снижения уровня метаболита М1 в плазме ниже чем 0,02 мг/л (0,02 мкг/мл). Полагают, на основании данных исследований у животных, что при концентрации активного метаболита М1 в плазме человека ниже чем 0,02 мг/л (0,02 мкг/мл) риск минимизирован.
Категория действия на плод по FDA. X.
Кормление грудью. Лефлуномид нельзя применять при кормлении грудью. Неизвестно, выделяется ли лефлуномид с грудным молоком. Поскольку многие ЛС проникают в молоко кормящих женщин, существует вероятность возникновения под влиянием лефлуномида серьезных неблагоприятных реакций у детей, находящихся на грудном вскармливании. Однако решение о начале лечения лефлуномидом или продолжении грудного вскармливания должно приниматься с учетом важности препарата для матери.
При воздействии на самок-крыс лефлуномидом в дозе 1,25 мг/кг, начиная за 14 дней до скрещивания и кончая завершением лактации, наблюдается снижение выживаемости потомства более чем на 90%. Уровень системной экспозиции при дозе 1,25 мг/кг был около 1/100 уровня экспозиции у человека, основанной на AUC.
Применение у женщин детородного возраста. Адекватных и строго контролированных исследований по оценке действия лефлуномида у беременных женщин проведено не было. Однако основываясь на данных исследований на животных, лефлуномид может повышать риск гибели плода при приеме в период беременности. Женщинам детородного возраста нельзя начинать лечение лефлуномидом до исключения беременности и (или) подтверждения использования надежной контрацепции. Перед началом лечения лефлуномидом пациентки должны быть в полном объеме проконсультированы о потенциальной серьезной опасности для плода.
Пациентки должны быть предупреждены, что при любой задержке наступления менструации или другой причине, предполагающей беременность, они должны немедленно известить врача для прохождения теста на беременность, и, при положительном результате, врач и пациентка должны обсудить риск для беременности. Вероятно, что быстрое снижение уровня активного метаболита в крови при использовании методики выведения препарата, при первой задержке менструации, сможет снизить риск применения лефлуномида для плода.
Всем женщинам детородного возраста после прекращения лечения лефлуномидом рекомендуется пройти процедуру выведения препарата. Женщинам, проходящим лечение лефлуномидом и желающим забеременеть, необходимо прервать прием препарата и пройти процедуру его выведения, включающую подтверждение снижения уровня метаболита М1 в плазме ниже чем 0,02 мг/л (0,02 мкг/мл). Полагают, на основании данных исследований у животных, что при концентрации активного метаболита М1 в плазме человека ниже чем 0,02 мг/л (0,02 мкг/мл) риск минимизирован.
Категория действия на плод по FDA. X.
Кормление грудью. Лефлуномид нельзя применять при кормлении грудью. Неизвестно, выделяется ли лефлуномид с грудным молоком. Поскольку многие ЛС проникают в молоко кормящих женщин, существует вероятность возникновения под влиянием лефлуномида серьезных неблагоприятных реакций у детей, находящихся на грудном вскармливании. Однако решение о начале лечения лефлуномидом или продолжении грудного вскармливания должно приниматься с учетом важности препарата для матери.
Method of drug use and dosage
Внутрь. Нагрузочная доза. Для быстрого достижения необходимого терапевтического уровня М1 в плазме крови, в начале терапии лефлуномидом необходимо использование нагрузочной дозы ЛС. В дальнейшем, в связи с длительным T1/2 из плазмы М1 и рекомендованным интервалом дозирования (24 ч), достаточно проведения терапии меньшими дозами. Рекомендуется инициировать терапию лефлуномидом нагрузочной дозировкой по 100 мг в день в течение 3 дней.
Поддерживающая терапия. Пациентам с РА для лечения рекомендуется ежедневная доза не более 20 мг. У небольшого числа пациентов (n=104), получавших 25 мг/день, было выявлено больше побочных реакций: облысение, потеря веса, подъем активности печеночных ферментов. Дозы выше 20 мг в день не рекомендуются. Если дозировка в 20 мг/день не переносится клинически хорошо, ее можно снизить до 10 мг в день. Уровень активности ферментов печени следует контролировать и, при необходимости, проводить коррекцию дозировки ( см «Ограничения к применению. Гепатотоксичность»). В связи с пролонгированным временем полужизни активного метаболита лефлуномида, пациенты должны тщательно наблюдаться после снижения дозы, может пройти несколько недель до снижения уровней метаболита.
Поддерживающая терапия. Пациентам с РА для лечения рекомендуется ежедневная доза не более 20 мг. У небольшого числа пациентов (n=104), получавших 25 мг/день, было выявлено больше побочных реакций: облысение, потеря веса, подъем активности печеночных ферментов. Дозы выше 20 мг в день не рекомендуются. Если дозировка в 20 мг/день не переносится клинически хорошо, ее можно снизить до 10 мг в день. Уровень активности ферментов печени следует контролировать и, при необходимости, проводить коррекцию дозировки ( см «Ограничения к применению. Гепатотоксичность»). В связи с пролонгированным временем полужизни активного метаболита лефлуномида, пациенты должны тщательно наблюдаться после снижения дозы, может пройти несколько недель до снижения уровней метаболита.
Side effects
Побочные реакции, связанные с применением лефлуномида при лечении РА, включают: диарею, увеличение активности печеночных ферментов (АЛТ и АСТ), алопецию и сыпь. Ниже представлены побочные реакции, выявленные в ходе контролируемых испытаний (независимо от причинности) и наблюдавшиеся с частотой ?3% хотя бы в одной из исследованных групп (табл. 4).
Таблица 4.
Побочные действия лефлуномида.
Все пациенты группы US301 принимали фолиевую кислоту; в группе MN301 фолиевую кислоту не принимали; только 10% пациентов группы MN302 принимали фолиевую кислоту.
1 - включая все контролируемые и не контролируемые испытания с лефлуномидом.
2 - гипертензия, как состояние, возникшее до начала испытаний, присутствовала во всех группах по исследованию действия лефлуномида в III фазе испытаний. Анализ случаев вновь возникшей гипертензии показывает отсутствие различий среди исследуемых групп.
В дополнение представлены побочные реакции, наблюдавшиеся у пациентов с РА, проходивших лечение лефлуномидом в контролируемых клинических испытаниях, с частотой от 1% до <3% .
Организм в целом. Абсцесс. Киста. Лихорадка. Грыжа. Недомогание. Боль. Боль в шее. Тазовая боль.
Сердечно-сосудистая система. Стенокардия. Мигрень. Сердцебиение. Тахикардия. Варикоз вен. Васкулит. Вазодилатация.
Желудочно-кишечная система. Желчнокаменная болезнь. Колит. Запор. Эзофагит. Метеоризм. Гастрит. Гингивит. Мелена. Кандидоз слизистой оболочки полости рта. Фарингит. Увеличение слюнных желез. Стоматит (или афтозный стоматит). Заболевания зубов.
Эндокринная система. Сахарный диабет, гипертиреоз.
Кроветворная и лимфатическая системы. Анемия (включая железодефицитную анемию), экхимоз.
Метаболизм. Повышение активности креатининфосфокиназы, гипергликемия, гиперлипидемия, периферический отек.
Скелетно-мышечная система. Артроз. Некроз кости. Костная боль. Бурсит. Мышечные судороги. Миалгия. Разрыв сухожилия.
Нервная система. Тревога. Депрессия. Сухость во рту. Инсомния. Невралгия. Неврит. Нарушение сна. Повышенная потливость. Головокружение.
Респираторная система. Астма, одышка, носовое кровотечение, заболевания легких.
Кожа и придатки кожи. Угри. Контактный дерматит. Грибковый дерматит. Изменение окраски волос и цвета кожи. Гематома. Простой герпес. Опоясывающий герпес. Папулезная сыпь. Заболевания ногтей. Кожные заболевания. Кожные узелки. Подкожные узелки. Язвы кожи.
Органы чувств. Нечеткость зрения, катаракта, конъюнктивит, заболевания глаз, извращение вкуса.
Мочеполовая система. Альбуминурия. Цистит. Дизурия. Гематурия. Менструальные расстройства. Заболевания предстательной железы. Учащение мочеиспускания. Вагинальный кандидоз.
Еще более редкие побочные реакции. Наблюдавшиеся в клинических испытаниях. Включали следующие: 1 случай анафилактической реакции. Произошедший в период исследований в Фазе II во время повторного начала испытания препарата после прерывания лечения из-за появления сыпи. Крапивница. Эозинофилия. Транзиторная тромбоцитопения (редко). Лейкопения <2000 клеток/мм3 (редко). В постмаркетинговых исследованиях наблюдались отдельные случаи панцитопении, синдрома Стивенса - Джонсона, токсического эпидермального некролиза и многоформной эритемы.
Таблица 4.
Побочные действия лефлуномида.
| Системы организма | Частота побочных реакций, % | ||||||
| Все исследования | Плацебо-контролируемые исследования | Плацебо-неконтролируемые исследования | |||||
| MN301 и US301 | MN 302 | ||||||
| Лефлуномид, n=13391 | Лефлуномид, n=315 | Плацебо, n=210 | Сульфасалазин, n=133 | Метотрексат, n=182 | Лефлуномид, n=501 | Метотрексат, n=498 | |
| Организм в целом | |||||||
| Аллергические реакции | 2 | 5 | 2 | 0 | 6 | 1 | 2 |
| Астения | 3 | 6 | 4 | 5 | 6 | 3 | 3 |
| Гриппозный синдром | 2 | 4 | 2 | 0 | 7 | 0 | 0 |
| Инфекции | 4 | 0 | 0 | 0 | 0 | 0 | 0 |
| Случайная травма | 5 | 7 | 5 | 3 | 11 | 6 | 7 |
| Боль | 2 | 4 | 2 | 2 | 5 | 1 | <1 |
| Боль в животе | 6 | 5 | 4 | 4 | 8 | 6 | 4 |
| Боль в спине | 5 | 6 | 3 | 4 | 9 | 8 | 7 |
| Сердечно-сосудистая | |||||||
| Гипертензия2 | 10 | 9 | 4 | 4 | 3 | 10 | 4 |
| Боль в груди | 2 | 4 | 2 | 2 | 4 | 1 | 2 |
| Желудочно-кишечная | |||||||
| Анорексия | 3 | 3 | 2 | 5 | 2 | 3 | 3 |
| Диарея | 17 | 27 | 12 | 10 | 20 | 22 | 10 |
| Диспепсия | 5 | 10 | 10 | 9 | 13 | 6 | 7 |
| Гастроэнтерит | 3 | 1 | 1 | 0 | 6 | 3 | 3 |
| Увеличение печени | 5 | 10 | 2 | 4 | 10 | 6 | 17 |
| Тошнота | 9 | 13 | 11 | 19 | 18 | 13 | 18 |
| Гастроинтестинальная/абдоминальная боль | 5 | 6 | 4 | 7 | 8 | 8 | 8 |
| Язвы во рту | 3 | 5 | 4 | 3 | 10 | 3 | 6 |
| Рвота | 3 | 5 | 4 | 4 | 3 | 3 | 3 |
| Метаболизм | |||||||
| Гипокалиемия | 1 | 3 | 1 | 1 | 1 | 1 | <1 |
| Снижение веса | 4 | 2 | 1 | 2 | 0 | 2 | 2 |
| Скелетно-мышечная | |||||||
| Артралгия | 1 | 4 | 3 | 0 | 9 | <1 | 1 |
| Судороги ног | 1 | 4 | 2 | 2 | 6 | 0 | 0 |
| Заболевания суставов | 4 | 2 | 2 | 2 | 2 | 8 | 6 |
| Синовит | 2 | <1 | 1 | 0 | 2 | 4 | 2 |
| Тендосиновит | 3 | 2 | 0 | 1 | 2 | 5 | 1 |
| Нервная | |||||||
| Головокружение | 4 | 5 | 3 | 6 | 5 | 7 | 6 |
| Головная боль | 7 | 13 | 11 | 12 | 21 | 10 | 8 |
| Парестезия | 2 | 3 | 1 | 1 | 2 | 4 | 3 |
| Респираторная | |||||||
| Бронхит | 7 | 5 | 2 | 4 | 7 | 8 | 7 |
| Усиление кашля | 3 | 4 | 5 | 3 | 6 | 5 | 7 |
| Респираторные инфекции | 15 | 21 | 21 | 20 | 32 | 27 | 25 |
| Фарингит | 3 | 2 | 1 | 2 | 1 | 3 | 3 |
| Пневмония | 2 | 3 | 0 | 0 | 1 | 2 | 2 |
| Ринит | 2 | 5 | 2 | 4 | 3 | 2 | 2 |
| Синусит | 2 | 5 | 5 | 0 | 10 | 1 | 1 |
| Кожа и придатки (кожи) | |||||||
| Облысение | 10 | 9 | 1 | 6 | 6 | 17 | 10 |
| Экзема | 2 | 1 | 1 | 1 | 1 | 3 | 2 |
| Зуд | 4 | 5 | 2 | 3 | 2 | 6 | 2 |
| Сыпь | 10 | 12 | 7 | 11 | 9 | 11 | 10 |
| Сухость кожи | 2 | 3 | 2 | 2 | 0 | 3 | 1 |
| Урогенитальная | |||||||
| Инфекции мочевыводящих путей | 5 | 5 | 7 | 4 | 2 | 5 | 6 |
Все пациенты группы US301 принимали фолиевую кислоту; в группе MN301 фолиевую кислоту не принимали; только 10% пациентов группы MN302 принимали фолиевую кислоту.
1 - включая все контролируемые и не контролируемые испытания с лефлуномидом.
2 - гипертензия, как состояние, возникшее до начала испытаний, присутствовала во всех группах по исследованию действия лефлуномида в III фазе испытаний. Анализ случаев вновь возникшей гипертензии показывает отсутствие различий среди исследуемых групп.
В дополнение представлены побочные реакции, наблюдавшиеся у пациентов с РА, проходивших лечение лефлуномидом в контролируемых клинических испытаниях, с частотой от 1% до <3% .
Организм в целом. Абсцесс. Киста. Лихорадка. Грыжа. Недомогание. Боль. Боль в шее. Тазовая боль.
Сердечно-сосудистая система. Стенокардия. Мигрень. Сердцебиение. Тахикардия. Варикоз вен. Васкулит. Вазодилатация.
Желудочно-кишечная система. Желчнокаменная болезнь. Колит. Запор. Эзофагит. Метеоризм. Гастрит. Гингивит. Мелена. Кандидоз слизистой оболочки полости рта. Фарингит. Увеличение слюнных желез. Стоматит (или афтозный стоматит). Заболевания зубов.
Эндокринная система. Сахарный диабет, гипертиреоз.
Кроветворная и лимфатическая системы. Анемия (включая железодефицитную анемию), экхимоз.
Метаболизм. Повышение активности креатининфосфокиназы, гипергликемия, гиперлипидемия, периферический отек.
Скелетно-мышечная система. Артроз. Некроз кости. Костная боль. Бурсит. Мышечные судороги. Миалгия. Разрыв сухожилия.
Нервная система. Тревога. Депрессия. Сухость во рту. Инсомния. Невралгия. Неврит. Нарушение сна. Повышенная потливость. Головокружение.
Респираторная система. Астма, одышка, носовое кровотечение, заболевания легких.
Кожа и придатки кожи. Угри. Контактный дерматит. Грибковый дерматит. Изменение окраски волос и цвета кожи. Гематома. Простой герпес. Опоясывающий герпес. Папулезная сыпь. Заболевания ногтей. Кожные заболевания. Кожные узелки. Подкожные узелки. Язвы кожи.
Органы чувств. Нечеткость зрения, катаракта, конъюнктивит, заболевания глаз, извращение вкуса.
Мочеполовая система. Альбуминурия. Цистит. Дизурия. Гематурия. Менструальные расстройства. Заболевания предстательной железы. Учащение мочеиспускания. Вагинальный кандидоз.
Еще более редкие побочные реакции. Наблюдавшиеся в клинических испытаниях. Включали следующие: 1 случай анафилактической реакции. Произошедший в период исследований в Фазе II во время повторного начала испытания препарата после прерывания лечения из-за появления сыпи. Крапивница. Эозинофилия. Транзиторная тромбоцитопения (редко). Лейкопения <2000 клеток/мм3 (редко). В постмаркетинговых исследованиях наблюдались отдельные случаи панцитопении, синдрома Стивенса - Джонсона, токсического эпидермального некролиза и многоформной эритемы.
Interaction
В in vivo исследованиях по взаимодействию ЛС выявлено несущественное взаимодействие между лефлуномидом и трехфазными контрацептивами для внутреннего применения, и циметидином.
In vitro исследования связывания ЛС с белками показали отсутствие воздействия варфарина на связывание белками активного метаболита М1. В то же время было показано, что М1 увеличивает на 13-50% содержание свободных фракций диклофенака, ибупрофена и толбутамида в концентрациях клинического диапазона. In vitro исследования метаболизма лефлуномида показали, что М1 ингибирует сYP2C9, который отвечает за метаболизм НПВС. Показано, что М1 ингибирует образование 4-гидроксидиклофенака из диклофенака in vitro. Клиническая значимость этих данных не известна, однако в клинических испытаниях широко использовалось сопутствующее применение НПВС и подобных эффектов не наблюдалось.
Прием колестирамина или активированного угля пациентами (n=13) и добровольцами (n=96) приводит к быстрому и достоверному снижению в плазме М1 ( см «Меры предосторожности»).
Метотрексат. У 30 пациентов совместный прием лефлуномида (100 мг/день в течение 2 дней, далее - 10-20 мг/день) и метотрексата (10-25 мг/нед с фолиевой кислотой) показал отсутствие фармакокинетического взаимодействия между препаратами. Однако совместный прием увеличивает риск гепатотоксичности.
Гепатотоксичные препараты. Возрастание побочных эффектов может проявиться при совместном приеме лефлуномида и гепатотоксичных соединений. Это необходимо принять во внимание, когда такие препараты применяются сразу после лечения лефлуномидом без предварительного использования процедуры выведения лефлуномида. В небольшом исследовании (n=30) при совместном применении лефлуномида и метотрексата у 5 из 30 пациентов наблюдались 2-3-кратные подъемы активности ферментов печени. Эти подъемы снижались: в 2-х случаях - при продолжении приема обоих препаратов, в 3-х случаях - после отмены лефлуномида. Более чем 3-кратное увеличение наблюдалось у других 5 пациентов. Эти подъемы снизились: у 2-х пациентов - при продолжении приема обоих препаратов, у 3-х - после отмены лефлуномида.
Рифампицин. При совместном приеме однократной дозы лефлуномида и многократных доз рифампицина уровни М1 повышались приблизительно на 40% по сравнению с их значением без приема рифампицина. Поэтому в связи с возможностью повышения уровней лефлуномида при его многократном применении, при совместном использовании с рифампицином, необходимо проявлять осторожность.
In vitro исследования связывания ЛС с белками показали отсутствие воздействия варфарина на связывание белками активного метаболита М1. В то же время было показано, что М1 увеличивает на 13-50% содержание свободных фракций диклофенака, ибупрофена и толбутамида в концентрациях клинического диапазона. In vitro исследования метаболизма лефлуномида показали, что М1 ингибирует сYP2C9, который отвечает за метаболизм НПВС. Показано, что М1 ингибирует образование 4-гидроксидиклофенака из диклофенака in vitro. Клиническая значимость этих данных не известна, однако в клинических испытаниях широко использовалось сопутствующее применение НПВС и подобных эффектов не наблюдалось.
Прием колестирамина или активированного угля пациентами (n=13) и добровольцами (n=96) приводит к быстрому и достоверному снижению в плазме М1 ( см «Меры предосторожности»).
Метотрексат. У 30 пациентов совместный прием лефлуномида (100 мг/день в течение 2 дней, далее - 10-20 мг/день) и метотрексата (10-25 мг/нед с фолиевой кислотой) показал отсутствие фармакокинетического взаимодействия между препаратами. Однако совместный прием увеличивает риск гепатотоксичности.
Гепатотоксичные препараты. Возрастание побочных эффектов может проявиться при совместном приеме лефлуномида и гепатотоксичных соединений. Это необходимо принять во внимание, когда такие препараты применяются сразу после лечения лефлуномидом без предварительного использования процедуры выведения лефлуномида. В небольшом исследовании (n=30) при совместном применении лефлуномида и метотрексата у 5 из 30 пациентов наблюдались 2-3-кратные подъемы активности ферментов печени. Эти подъемы снижались: в 2-х случаях - при продолжении приема обоих препаратов, в 3-х случаях - после отмены лефлуномида. Более чем 3-кратное увеличение наблюдалось у других 5 пациентов. Эти подъемы снизились: у 2-х пациентов - при продолжении приема обоих препаратов, у 3-х - после отмены лефлуномида.
Рифампицин. При совместном приеме однократной дозы лефлуномида и многократных доз рифампицина уровни М1 повышались приблизительно на 40% по сравнению с их значением без приема рифампицина. Поэтому в связи с возможностью повышения уровней лефлуномида при его многократном применении, при совместном использовании с рифампицином, необходимо проявлять осторожность.
Overdose
Данные, касающиеся передозировки лефлуномида при применении его у людей, отсутствуют. В острых токсикологических экспериментах на мышах и крысах минимальными токсическими дозами при пероральном применении лефлуномида были 200-500 мг/кг и 100 мг/кг. Соответственно (приблизительно более чем в 350 раз выше максимальной рекомендуемой человеку дозы). В случае значительной передозировки или проявления токсичности, для ускорения выведения препарата, рекомендуется прием колестирамина или активированного угля ( см «Меры предосторожности. Необходимость в выведении препарата»).
Application precautions
Необходимость в выведении препарата. Активный метаболит лефлуномида медленно выводится из плазмы. В случаях проявления высокой токсичности лефлуномида и/или гиперчувствительности, необходимо применение методики выведения препарата для более быстрого снижения концентрации ЛС в плазме после прекращения терапии лефлуномидом. Пролонгированный прием колестирамина или активированного угля является необходимым для достижения быстрого и достаточного выведения, длительность приема может изменяться в зависимости от клинического статуса пациента. Колестирамин, принимаемый внутрь в дозировке 8 г 3 раза в день в течение 24 ч тремя здоровыми добровольцами, снижал плазматические уровни М1 в течение 24 ч приблизительно на 40%, а в течение 48 ч - на 49-65%. Было показано, что прием активированного угля (порошок в виде суспензии) внутрь или через желудочный зонд (по 50 г каждые 6 ч в течение 24 ч) приводит к снижению плазматических концентраций активного метаболита М1 на 37% за 24 ч и на 48% за 48 Эти процедуры выведения препарата могут быть повторены в случае клинической необходимости.
Почечная недостаточность. В исследованиях с однократным приемом лефлуномида было показано, что у пациентов, прошедших процедуру гемодиализа, концентрация свободного М1 в плазме удваивается. Однако нет клинических данных о применении лефлуномида у больных с почечными нарушениями. Поэтому следует с осторожностью применять препарат у данной группы больных.
Вакцинация. Клинические данные об эффективности и безопасности вакцинации во время лечения лефлуномидом отсутствуют. Однако вакцинация живыми вакцинами не рекомендуется. Необходимо учитывать длительность времени полужизни лефлуномида при намерении введения живой вакцины после прекращения терапии лефлуномидом.
Лабораторные исследования. Уровень активности АЛТ является основным диагностическим показателем гепатотоксичности и должен контролироваться ежемесячно, а затем, при стабилизации, определяться по индивидуальной клинической ситуации.
У пациентов с повышенным риском проявления гематотоксичности должен быть гарантирован более тщательный контроль, включая гематологический ( см «Ограничения к применению. Возможность иммунодепрессии»).
В связи со специфическим действием на щеточную каемку почечных проксимальных канальцев, лефлуномид вызывает ускорение выведения мочевой кислоты (урикозурический эффект). У некоторых пациентов отдельно наблюдается гипофосфатурия. Эти эффекты совместно не наблюдаются и не являются альтернативными друг другу.
Применение у мужчин. Доступная информация не дает оснований для предположения, что применение лефлуномида повышает риск зависимой от мужчины фетальной токсичности. Экспериментов на животных, посвященных оценке этого специфического риска, не проводилось. Для минимизации любого возможного риска мужчины, желающие стать отцами, должны прервать лечение лефлуномидом и принимать колестирамин внутрь по 8 г 3 раза ежедневно в течение 11 дней.
Использование в педиатрии. Безопасность и эффективность лефлуномида при применении у детей не исследована. Использование лефлуномида пациентами моложе 18 лет не рекомендуется.
Гериатрия. У пациентов старше 65 лет коррекция дозирования лефлуномида не требуется.
Возможность злоупотребления и зависимости от лефлуномида не выявлена.
Почечная недостаточность. В исследованиях с однократным приемом лефлуномида было показано, что у пациентов, прошедших процедуру гемодиализа, концентрация свободного М1 в плазме удваивается. Однако нет клинических данных о применении лефлуномида у больных с почечными нарушениями. Поэтому следует с осторожностью применять препарат у данной группы больных.
Вакцинация. Клинические данные об эффективности и безопасности вакцинации во время лечения лефлуномидом отсутствуют. Однако вакцинация живыми вакцинами не рекомендуется. Необходимо учитывать длительность времени полужизни лефлуномида при намерении введения живой вакцины после прекращения терапии лефлуномидом.
Лабораторные исследования. Уровень активности АЛТ является основным диагностическим показателем гепатотоксичности и должен контролироваться ежемесячно, а затем, при стабилизации, определяться по индивидуальной клинической ситуации.
У пациентов с повышенным риском проявления гематотоксичности должен быть гарантирован более тщательный контроль, включая гематологический ( см «Ограничения к применению. Возможность иммунодепрессии»).
В связи со специфическим действием на щеточную каемку почечных проксимальных канальцев, лефлуномид вызывает ускорение выведения мочевой кислоты (урикозурический эффект). У некоторых пациентов отдельно наблюдается гипофосфатурия. Эти эффекты совместно не наблюдаются и не являются альтернативными друг другу.
Применение у мужчин. Доступная информация не дает оснований для предположения, что применение лефлуномида повышает риск зависимой от мужчины фетальной токсичности. Экспериментов на животных, посвященных оценке этого специфического риска, не проводилось. Для минимизации любого возможного риска мужчины, желающие стать отцами, должны прервать лечение лефлуномидом и принимать колестирамин внутрь по 8 г 3 раза ежедневно в течение 11 дней.
Использование в педиатрии. Безопасность и эффективность лефлуномида при применении у детей не исследована. Использование лефлуномида пациентами моложе 18 лет не рекомендуется.
Гериатрия. У пациентов старше 65 лет коррекция дозирования лефлуномида не требуется.
Возможность злоупотребления и зависимости от лефлуномида не выявлена.
Special instructions
Информация для пациентов. Необходимо обсудить с пациентками - женщинами детородного возраста - вероятность повышенного риска врожденных дефектов у плода. Женщинам при посещении врача необходимо объяснять, что они будут находиться в группе повышенного риска рождения ребенка с врожденными дефектами, если начинать принимать лефлуномид во время беременности, забеременеть во время прохождения курса лечения или до прерывания лечения и проведения процедуры выведения препарата. Мужчины, желающие стать отцами, должны прервать лечение лефлуномидом.
Пациенты должны быть предупреждены о возможности редких, но серьезных кожных реакций, о необходимости безотлагательного контакта с врачом в случае появления кожной сыпи или поражения слизистых оболочек.
Пациенты должны быть извещены о возможных гепатотоксических эффектах лефлуномида и необходимости контроля активности ферментов печени.
Пациенты, которые получают иммунодепрессивную терапию совместно с приемом лефлуномида, или закончили эту терапию перед лечением лефлуномидом, или имеют в анамнезе существенную гематологическую патологию, должны быть извещены о возможном развитии панцитопении и необходимости частого гематологического контроля. Они должны быть проинструктированы о незамедлительном извещении врача в случае обнаружения симптомов панцитопении (легкое образование подкожных гематом, предрасположенность к инфекциям, бледность и повышенная утомляемость).
Пациенты должны быть предупреждены о возможности редких, но серьезных кожных реакций, о необходимости безотлагательного контакта с врачом в случае появления кожной сыпи или поражения слизистых оболочек.
Пациенты должны быть извещены о возможных гепатотоксических эффектах лефлуномида и необходимости контроля активности ферментов печени.
Пациенты, которые получают иммунодепрессивную терапию совместно с приемом лефлуномида, или закончили эту терапию перед лечением лефлуномидом, или имеют в анамнезе существенную гематологическую патологию, должны быть извещены о возможном развитии панцитопении и необходимости частого гематологического контроля. Они должны быть проинструктированы о незамедлительном извещении врача в случае обнаружения симптомов панцитопении (легкое образование подкожных гематом, предрасположенность к инфекциям, бледность и повышенная утомляемость).
References
1. Physicians Desk Reference. 59-th ed. Thomson PDR. 2005. P. 693-699.
2. Sharp J.T. Scoring radiographic abnormalities in rheumatoid arthritis//Radiologic сlinics of North America. 1996. V. 34. P. 233-241.
Описание проверено.
•.
Крылов Юрий Федорович.
(фармаколог, доктор медицинских наук, профессор, академик Международной академии информатизации).
Опыт работы: более 35 лет.
2. Sharp J.T. Scoring radiographic abnormalities in rheumatoid arthritis//Radiologic сlinics of North America. 1996. V. 34. P. 233-241.
Описание проверено.
•.
Крылов Юрий Федорович.
(фармаколог, доктор медицинских наук, профессор, академик Международной академии информатизации).
Опыт работы: более 35 лет.
Included in the composition
- 2.7-10€ Leflunomide Canon
- 6.5-10€ Leflunomide (6 firms)
- 4.1-13.1€ Arava (4 firms)
- 9.4-11€ Ehlafra (2 firms)
- 10.7-10.9€ Ralef
- 18.9-19.6€ Leflaid (Технология лекарств)
- — Lefomide (3 firms)
- — Арресто (Simpex Pharma)