ICD-10 codes
Description
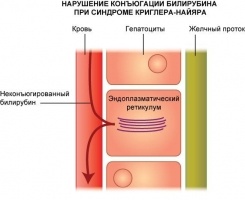
Синдром Криглера. Найяра - генетическое заболевание из класса ферментопатий, характеризующееся нарушением одного из звеньев процесса обезвреживания и выведения билирубина - конъюгации. Симптомами этого состояния являются желтуха печеночного генеза и тяжелые неврологические нарушения, которые могут привести к летальному исходу еще в младенческом возрасте. Диагностика синдрома Криглера-Найяра производится посредством биохимических проб и определения уровня неконъюгированного билирубина в плазме крови, а также молекулярно-генетическими методиками. Специфического лечения заболевания не существует (за исключением трансплантации печени), терапия сводится к увеличению разрушения и элиминации билирубина из организма (гемосорбция, фототерапия, плазмаферез, прием барбитуратов).
Additional facts
Синдром Криглера-Найяра - тяжелое генетическое заболевание, характеризующееся нарушением связывания билирубина с глюкуроновой кислотой, что является ключевым этапом его обезвреживания и выведения из организма. Впервые это заболевание было описано в 1952 году двумя американскими педиатрами - Джоном Криглером и Виктором Найяром. Дальнейшее изучение синдрома Криглера-Найяра показало, что это состояние имеет генетическую природу и аутосомно-рецессивный характер наследования, кроме того, удалось выявить две клинические разновидности данной патологии. Заболевание достаточно редкое, поэтому точные цифры встречаемости не определены - большинство исследователей полагает, что она находится на уровне 1:1 000 000. Половое распределение больных синдромом Криглера-Найяра не имеет особенностей, заболевание с одинаковой частотой поражает как мальчиков, так и девочек. В лечении этого состояния крайне важна ранняя (в идеале - пренатальная) диагностика, так как от своевременности начатой терапии очень сильно зависят прогноз заболевания и качество жизни больного.
Reasons
Синдром Криглера-Найяра относят к классу ферментопатий (по другой классификации - к группе неконъюгированных гипербилирубинемий), причина этого заболевания кроется в недостаточности уридиндифосфатглюкуронидазы 1, функцией которой является связывание билирубина с двумя молекулами глюкуроновой кислоты. В итоге этого биохимического процесса билирубин становится способным растворяться в воде, выводиться в составе желчи и, главное, значительно падает его токсичность. При синдроме Криглера-Найяра этот процесс резко замедлен или не происходит совсем, вследствие чего возникает задержка элиминации билирубина из организма и его накопление. Билирубин обладает выраженной нейротоксичностью, при повышении концентрации в крови это вещество начинает откладываться в тканях кожных покровов и слизистых оболочек, приводя к развитию желтухи. Когда концентрация билирубина превышает определенный порог, соединение начинает проникать через гематоэнцефалический барьер в головной мозг, приводя к тяжелой энцефалопатии (особенно повреждаются базальные ядра). При отсутствии лечения больные синдромом Криглера-Найяра погибают от многочисленных неврологических расстройств и нарастающей печеночной комы.
Причиной низкой активности уридиндифосфатглюкуронидазы являются мутации гена UGT1A1, который располагается на 2-й хромосоме, отвечает за аминокислотную последовательность и выделение этого фермента. Помимо синдрома Криглера-Найяра дефекты этого гена могут приводить к ряду других нарушений билирубинового обмена наследственного характера - синдрому Жильбера, транзиторной неонатальной билирубинемии семейного типа. Механизм наследования мутаций гена UGT1A1 при синдроме Криглера-Найяра аутосомно-рецессивный. При этом описано несколько вариантов возможного повреждения этого гена, которые приводят к разному течению данного заболевания.
Причиной низкой активности уридиндифосфатглюкуронидазы являются мутации гена UGT1A1, который располагается на 2-й хромосоме, отвечает за аминокислотную последовательность и выделение этого фермента. Помимо синдрома Криглера-Найяра дефекты этого гена могут приводить к ряду других нарушений билирубинового обмена наследственного характера - синдрому Жильбера, транзиторной неонатальной билирубинемии семейного типа. Механизм наследования мутаций гена UGT1A1 при синдроме Криглера-Найяра аутосомно-рецессивный. При этом описано несколько вариантов возможного повреждения этого гена, которые приводят к разному течению данного заболевания.
Symptoms
В настоящее время описаны две основные клинические формы синдрома Криглера-Найяра, в основном различающиеся между собой тяжестью проявлений и прогнозом заболевания. Это обусловлено типом генетического дефекта в UGT1A1. Первый тип заболевания (СКН-1) вызывается миссенс-мутациями, приводящими к появлению неполноценного фермента, имеющего сигнальную последовательность аминокислот, характерную для подвергающихся внутриклеточной утилизации белков. Таким образом, при этой форме дефект гена поражает кодирующие участки (экзоны), что вызывает развитие патологии у гомозигот. Вскоре после своего образования уридиндифосфатглюкуронидаза 1 разрушается и конъюгации билирубина не происходит совсем.
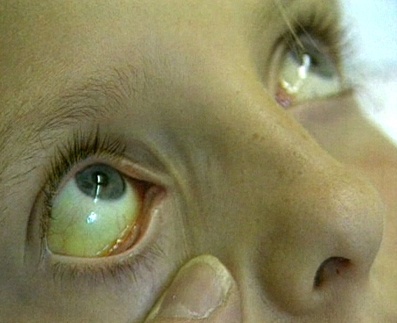
Синдром Криглера-Найяра 1-го типа характеризуется тяжелым и стремительным течением - первые признаки гипербилирубинемии в виде желтухи обнаруживаются уже через несколько часов после рождения. Со временем к ним присоединяются неврологические нарушения - нистагм, судорожные приступы, иногда возникает опистотонус. Желтуха сохраняется на протяжении всей жизни ребенка, его умственное развитие резко отстает от такового у сверстников, симптомы заболевания неуклонно нарастают даже при интенсивном лечении. Обычно больные синдромом Криглера-Найяра 1-го типа умирают на протяжении первого года жизни из-за интоксикации билирубином и поражения базальных подкорковых ядер (ядерная энцефалопатия).
Причиной синдрома Криглера-Найяра 2-го типа также являются миссенс-мутации гена UGT1A1, однако они могут возникать как в кодирующей последовательности (экзонах), так и в промоторе - участке, отвечающем за экспрессию данного гена. У большинства больных СКН-2 наблюдается наличие на одной хромосоме дефекта экзона, на другой - промотора, то есть, такие лица являются компаунд-гетерозиготами. Результатом нарушения является продукция дефектной формы фермента уридиндифосфатглюкуронидазы, которая не разрушается, но имеет пониженную (на уровне 20-25% от нормы) функциональную активность. Поэтому синдром Криглера-Найяра 2-го типа характеризуется менее тяжелой клинической картиной и более благоприятным прогнозом.
Ассоциированные симптомы: Гипербилирубинемия. Кал желтого цвета. Кал серовато-белого цвета. Судороги. Тяжесть в подреберье.
Синдром Криглера-Найяра 1-го типа характеризуется тяжелым и стремительным течением - первые признаки гипербилирубинемии в виде желтухи обнаруживаются уже через несколько часов после рождения. Со временем к ним присоединяются неврологические нарушения - нистагм, судорожные приступы, иногда возникает опистотонус. Желтуха сохраняется на протяжении всей жизни ребенка, его умственное развитие резко отстает от такового у сверстников, симптомы заболевания неуклонно нарастают даже при интенсивном лечении. Обычно больные синдромом Криглера-Найяра 1-го типа умирают на протяжении первого года жизни из-за интоксикации билирубином и поражения базальных подкорковых ядер (ядерная энцефалопатия).
Причиной синдрома Криглера-Найяра 2-го типа также являются миссенс-мутации гена UGT1A1, однако они могут возникать как в кодирующей последовательности (экзонах), так и в промоторе - участке, отвечающем за экспрессию данного гена. У большинства больных СКН-2 наблюдается наличие на одной хромосоме дефекта экзона, на другой - промотора, то есть, такие лица являются компаунд-гетерозиготами. Результатом нарушения является продукция дефектной формы фермента уридиндифосфатглюкуронидазы, которая не разрушается, но имеет пониженную (на уровне 20-25% от нормы) функциональную активность. Поэтому синдром Криглера-Найяра 2-го типа характеризуется менее тяжелой клинической картиной и более благоприятным прогнозом.
Ассоциированные симптомы: Гипербилирубинемия. Кал желтого цвета. Кал серовато-белого цвета. Судороги. Тяжесть в подреберье.
Classification
В первые месяцы и даже годы жизни больных синдром Криглера-Найяра этого типа нередко проявляется только незначительной желтухой, при отсутствии лечения к подростковому периоду могут развиваться неврологические отклонения. В ряде случаев, особенно при правильно назначенных терапевтических мероприятиях, никаких нарушений со стороны центральной нервной системы не возникает вовсе. Проявления желтухи различной степени выраженности у больных синдромом Криглера-Найяра 2-го типа могут сохраняться на протяжении всей жизни и нередко расцениваются как индикатор осложнений и ухудшения состояния пациента. С возрастом иногда появляется нистагм, могут регистрироваться судорожные припадки, однако течение и выраженность симптомов заболевания всецело зависят от качества лечения и выполнения рекомендаций специалистов.
Diagnostics
Диагностика синдрома Криглера-Найяра производится на основании данных общего осмотра ребенка, биохимических исследований крови, желчи и мочи, молекулярно-генетических анализов. При осмотре выявляется желтуха, возникшая в первые часы (при СКН-1) или месяцы (СКН-2) жизни, признаки неврологических нарушений (опистотонус, нистагм, длительное сохранение транзиторных рефлексов). У больных 2-м типом синдрома Криглера-Найяра неврологические расстройства могут регистрироваться во взрослом возрасте, тогда как у детей наблюдается только желтуха. Также с возрастом могут присоединяться такие проявления, как нейросенсорная глухота или хореоатетоз.
При биохимическом исследовании крови выявляется выраженная непрямая гипербилирубинемия (вплоть 200-350 мкмоль/л), отсутствие (при синдроме Криглера-Найяра 1-го типа) или резкое снижение концентрации прямого билирубина. Конъюгированная фракция этого соединения отсутствует в желчи при СКН-1 и присутствует в незначительных количествах при СКН-2. Фенобарбиталовая проба при синдроме Криглера-Найяра положительна только в случае наличия уридиндифосфатглюкуронидазы, то есть при СКН-2. Изучение концентрации неконъюгированного билирубина в моче показывает его увеличение. Молекулярно-генетическая диагностика синдрома Криглера-Найяра производится врачом-генетиком - он совершает прямое секвенирование последовательности гена UGT1A1 с целью выявления мутаций. При отягощенной по этому заболеванию наследственности у родителей может осуществляться пренатальная диагностика патологии.
При биохимическом исследовании крови выявляется выраженная непрямая гипербилирубинемия (вплоть 200-350 мкмоль/л), отсутствие (при синдроме Криглера-Найяра 1-го типа) или резкое снижение концентрации прямого билирубина. Конъюгированная фракция этого соединения отсутствует в желчи при СКН-1 и присутствует в незначительных количествах при СКН-2. Фенобарбиталовая проба при синдроме Криглера-Найяра положительна только в случае наличия уридиндифосфатглюкуронидазы, то есть при СКН-2. Изучение концентрации неконъюгированного билирубина в моче показывает его увеличение. Молекулярно-генетическая диагностика синдрома Криглера-Найяра производится врачом-генетиком - он совершает прямое секвенирование последовательности гена UGT1A1 с целью выявления мутаций. При отягощенной по этому заболеванию наследственности у родителей может осуществляться пренатальная диагностика патологии.
Differential diagnostics
Дифференциальный диагноз следует проводить с обычной транзиторной желтухой новорожденных и синдромом Жильбера.
Treatment
Специфического или этиотропного лечения синдрома Криглера-Найяра на сегодняшний день не существует, все терапевтические мероприятия назначаются для ускорения распада билирубина, его выведения из организма и защиты ЦНС. Особых отличий в терапии 1-го или 2-го типа заболевания нет (за исключением активизации микросомального окисления барбитуратами, которая не производится при 1-м типе), однако при СКН-1 лечение лишь незначительно оттягивает наступление летального исхода. Самым радикальным методом лечения синдрома Криглера-Найяра в настоящее время является операция по аллотрансплантации печени от родственника или генетически сходного донора - в этом органе происходит образование уридиндифосфатглюкуронидазы.
Синдром Криглера-Найяра 2-го типа лечат назначением умеренных доз барбитуратов для активации окисления билирубина и увеличения образования нужного фермента. Кроме того, показаны плазмаферез, гемосорбция, заместительное переливание крови - все эти процедуры направлены на удаление неконъюгированного билирубина из организма. Неплохие результаты у больных синдромом Криглера-Найяра дает фототерапия - облучение кожных покровов приводит к частичному разрушению билирубина и освобождению рецепторов тканей для новых порций этого токсина, что снижает его концентрацию в крови. Правильный питьевой режим и повышенное потребление жидкости ускоряет выведение токсина из организма, поэтому следует избегать обезвоживания. Необходим постоянный мониторинг уровня этого вещества в плазме крови, особенно опасным считается его количество свыше 300-340 мкмоль/л - при такой концентрации билирубин становится способным проникать через гематоэнцефалический барьер.
Синдром Криглера-Найяра 2-го типа лечат назначением умеренных доз барбитуратов для активации окисления билирубина и увеличения образования нужного фермента. Кроме того, показаны плазмаферез, гемосорбция, заместительное переливание крови - все эти процедуры направлены на удаление неконъюгированного билирубина из организма. Неплохие результаты у больных синдромом Криглера-Найяра дает фототерапия - облучение кожных покровов приводит к частичному разрушению билирубина и освобождению рецепторов тканей для новых порций этого токсина, что снижает его концентрацию в крови. Правильный питьевой режим и повышенное потребление жидкости ускоряет выведение токсина из организма, поэтому следует избегать обезвоживания. Необходим постоянный мониторинг уровня этого вещества в плазме крови, особенно опасным считается его количество свыше 300-340 мкмоль/л - при такой концентрации билирубин становится способным проникать через гематоэнцефалический барьер.