Другие названия и синонимы
Lipoidosis, Лизосомные болезни накопления липидов, Липидозы, Ретикулоэндотелиозы.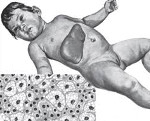
МКБ-10 коды
|
|
Описание
Наследственные заболевания, связанные с нарушением жирового обмена, отложением липидов и их метаболитов в различных органах и тканях. Общие клинические проявления представлены прогрессирующим нарушением интеллектуальных и двигательных функций, повреждением костей, кожи, центральной нервной системы, глаз и внутренних органов (печени, почек, селезенки). Диагноз основывается на лабораторных исследованиях активности фермента, количества токсического субстрата, наличия мутаций в генах. Лечение включает замену фермента, уменьшение количества субстрата и симптоматическую терапию.
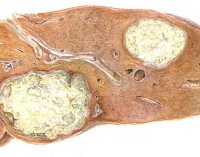
Дополнительные факты
Синонимами для липоидозов являются липидозы, ретикулоэндотелиоз, лизосомные заболевания накопления липидов. В эту группу входят сфингомиелиноз, болезнь Тея-Сакса, болезнь Гоше, семейная гиперлипидемия и некоторые другие заболевания. Общей чертой является патологическое накопление липидов в клетках организма в результате дефекта ферментных систем. Липоидозы относятся к группе редких (сиротских) заболеваний. Его распространенность очень низкая, так как некоторые типы патологий варьируются от 1:40 тысяч до 1: 1 миллиона или меньше. Общая частота составляет 1: 7000. Большинство липидозов имеют прогрессирующее течение, приводящее к инвалидности и ранней смерти.
Причины
Определяющим фактором в развитии липидоза является генетический дефект, который вызывает полную или частичную недостаточность лизосомальных ферментов, расщепляющих сложные липиды. Заболевания наследуются по аутосомно-рецессивному механизму. Это означает, что новорожденный болен, если он получает ген мутации от каждого родителя (имеет пару дефектных генов в аллеле). Если мутация передается только от матери или отца, ребенок является ее носителем и остается здоровым.
Исключительный механизм передачи дефектов при болезни Фабри. В отличие от других липидозов он наследуется от Х-хромосомного рецессивного типа. Пациенты-мужчины с гемизиготами больны и передают мутацию только дочерям. У девочек болезнь всегда проявляется при наличии двух рецессивных (измененных) генов. Иногда симптомы липидоза возникают у пациентов с дефектным геном, когда доминантный ген инактивирован (причины этого неясны).
Исключительный механизм передачи дефектов при болезни Фабри. В отличие от других липидозов он наследуется от Х-хромосомного рецессивного типа. Пациенты-мужчины с гемизиготами больны и передают мутацию только дочерям. У девочек болезнь всегда проявляется при наличии двух рецессивных (измененных) генов. Иногда симптомы липидоза возникают у пациентов с дефектным геном, когда доминантный ген инактивирован (причины этого неясны).
Патогенез
Патогенной основой большинства липоидозов является генетически обусловленный дефект фермента - болезнь фермента. В результате клеточные лизосомы накапливают липиды и интермедиаты в своем метаболизме, что приводит к постепенному увеличению органической дисфункции. При синдроме Вольмана определяется дефицит кислой этеразы, полный цикл метаболизма холестерина становится невозможным, содержание его сложных эфиров в лизосомах селезенки, печени, надпочечников, кишечника и костного мозга увеличивается. У пациентов с болезнью Гоше выработка бета-глюкозидазы снижается или полностью отсутствует. В печени, селезенке, костном мозге, накапливаются сфинголипидные продукты расщепления. Болезнь Нимана-Пика развивается на основе дефицита сфингомиелазы, характеризующейся увеличением количества сфингомиелина во многих клетках, в частности в гепатоцитах, нейронах. При идиотизме Тэя-Сакса возникает дефект в N-ацетилгексозаминидазе, и ганглиозиды накапливаются в мозге.
Классификация
Наследственные метаболические нарушения сложных липидов представлены группой заболеваний различного происхождения, которые на патогенетическом уровне связаны с патологией жирового обмена. Накопление сложных липидных соединений в клеточных лизосомах характерно для липоидоза. Различные типы заболеваний различаются в зависимости от того, какой липид не полностью расщеплен и откладывается в ткани:
При этом типе болезни полный распад гликолипидов - соединений, состоящих из углеводов и жирных кислот - невозможен. Гликолипидозы представлены цереброзидозами (сфинголипидоз Гоше, болезнь Крэбба), сульфатидозами (метахромотическая лейкодистрофия), церамидолигозидозами (болезнь Фабри, церамид-лактозидный липоидоз), ганглиозидозами (болезнь Сандхоффа, амиотра в раннем детстве).
В связи с патологией липидного обмена плазмы крови, на основе генетического дефекта ферментов или клеточных рецепторов. Плазменные липиды - жирные кислоты, триглицериды, холестерин. Липопротеинемии включают семейную гиперхолестеринемию, комбинированную семейную гиперлипидемию и семейную гиперлипидемию I-V типа.
Синоним имени - болезнь Нимана-Пика. С развитием этого заболевания в ретикулоэндотелиальных клетках содержание фосфолипидного соединения сфингомиелина увеличивается.
При этом типе болезни полный распад гликолипидов - соединений, состоящих из углеводов и жирных кислот - невозможен. Гликолипидозы представлены цереброзидозами (сфинголипидоз Гоше, болезнь Крэбба), сульфатидозами (метахромотическая лейкодистрофия), церамидолигозидозами (болезнь Фабри, церамид-лактозидный липоидоз), ганглиозидозами (болезнь Сандхоффа, амиотра в раннем детстве).
В связи с патологией липидного обмена плазмы крови, на основе генетического дефекта ферментов или клеточных рецепторов. Плазменные липиды - жирные кислоты, триглицериды, холестерин. Липопротеинемии включают семейную гиперхолестеринемию, комбинированную семейную гиперлипидемию и семейную гиперлипидемию I-V типа.
Синоним имени - болезнь Нимана-Пика. С развитием этого заболевания в ретикулоэндотелиальных клетках содержание фосфолипидного соединения сфингомиелина увеличивается.
Клиническая картина
Клиническая картина липидоза определяется особенностями участия в патологическом процессе органов и систем. Болезнь Гоше 1 типа поражает печень, селезенку, костный и костный мозг. Симптомы включают увеличение размера печени, ломкость костей, анемию, лейкопению и снижение свертываемости крови. Болезнь типа II развивается с преимущественным повреждением центральной нервной системы и печени. Наблюдаются судороги, гипертонус мышц, спастичность, умственная недостаточность и нарушения глотания. Функциональность миелиновой оболочки снижается у людей с галактозилцерамидным липидозом (болезнь Крэбба). Развиваются гипервозбудимость, рвота, судороги, задержка психомоторного развития с прогрессирующим снижением интеллекта и зрения.
При метахромотической лейкодистрофии миелиновая оболочка изменяется патологически. Основными симптомами являются гипотензия мышц рук и ног, снижение (отсутствие) сухожильных рефлексов, атаксия, атрофия зрительных нервов, нистагм, спастический тетрапарез, онемение, умственное и моторное недоразвитие. Клинические признаки болезни Андерсона Фабри разнообразны. Одним из наиболее распространенных являются невропатические боли (умеренные и слабые, на ступнях и кистях рук) и фабри-кризы - сильные жгучие боли в конечностях пароксизмальной природы. Существует также гипогидроз или ангидроз, непереносимость физических нагрузок, ангиокератома, потеря слуха, сердечно-сосудистые и почечные функции.
С развитием синдрома Сандхоффа определяются общая вялость, гипотензия мышц конечностей, затруднение сосания и глотания, прогрессирующая задержка двигательного и умственного развития, моторная слабость, шум в сердце, судороги, слепота, увеличение селезенки. При раннем детстве амавротический идиотизм поражает центральную нервную систему. К концу первого полугодия реакция детей на внешние сигналы (лица родственников, игрушки) ухудшается, моторные навыки теряются, познавательный и игровой интерес снижается. Зрение нарушено до слепоты. Судорожная форма судорог.
Первыми симптомами сфингомиелиноза являются вялость, малоподвижность, вялость, отказ от еды, рвота. Позже живот (гепатомегалия) увеличивается, конечности истончаются, кожа приобретает коричневатый оттенок, периоды торможения сменяются гипервозбуждением. Дети отстают в психофизическом развитии. Поставлен диагноз: умеренная гидроцефалия, гипертермия, спастический парез ног и рук, пароксизмальная асфиксия.
Проявления гиперлипопротеинемии обнаруживаются в возрасте до 10 лет. Все формы заболевания характеризуются отложениями подкожного жира в виде ксантом, а также болями в животе, панкреатитом, гепатоспленомегалией (накопление липидов в селезенке, печени). При сочетанной семейной гиперлипидемии и гиперлипидемии II типа возможен ранний атеросклероз. При гиперлипидемии IV и V типов, снижении чувствительности к глюкозе, ишемической болезни сердца.
Ассоциированные симптомы: Общая слабость. Гипербилирубинемия. Рвота. Снижение потоотделения. Судороги. Судороги в ногах. Тетрапарез. Хромота.
При метахромотической лейкодистрофии миелиновая оболочка изменяется патологически. Основными симптомами являются гипотензия мышц рук и ног, снижение (отсутствие) сухожильных рефлексов, атаксия, атрофия зрительных нервов, нистагм, спастический тетрапарез, онемение, умственное и моторное недоразвитие. Клинические признаки болезни Андерсона Фабри разнообразны. Одним из наиболее распространенных являются невропатические боли (умеренные и слабые, на ступнях и кистях рук) и фабри-кризы - сильные жгучие боли в конечностях пароксизмальной природы. Существует также гипогидроз или ангидроз, непереносимость физических нагрузок, ангиокератома, потеря слуха, сердечно-сосудистые и почечные функции.
С развитием синдрома Сандхоффа определяются общая вялость, гипотензия мышц конечностей, затруднение сосания и глотания, прогрессирующая задержка двигательного и умственного развития, моторная слабость, шум в сердце, судороги, слепота, увеличение селезенки. При раннем детстве амавротический идиотизм поражает центральную нервную систему. К концу первого полугодия реакция детей на внешние сигналы (лица родственников, игрушки) ухудшается, моторные навыки теряются, познавательный и игровой интерес снижается. Зрение нарушено до слепоты. Судорожная форма судорог.
Первыми симптомами сфингомиелиноза являются вялость, малоподвижность, вялость, отказ от еды, рвота. Позже живот (гепатомегалия) увеличивается, конечности истончаются, кожа приобретает коричневатый оттенок, периоды торможения сменяются гипервозбуждением. Дети отстают в психофизическом развитии. Поставлен диагноз: умеренная гидроцефалия, гипертермия, спастический парез ног и рук, пароксизмальная асфиксия.
Проявления гиперлипопротеинемии обнаруживаются в возрасте до 10 лет. Все формы заболевания характеризуются отложениями подкожного жира в виде ксантом, а также болями в животе, панкреатитом, гепатоспленомегалией (накопление липидов в селезенке, печени). При сочетанной семейной гиперлипидемии и гиперлипидемии II типа возможен ранний атеросклероз. При гиперлипидемии IV и V типов, снижении чувствительности к глюкозе, ишемической болезни сердца.
Ассоциированные симптомы: Общая слабость. Гипербилирубинемия. Рвота. Снижение потоотделения. Судороги. Судороги в ногах. Тетрапарез. Хромота.
|
|
Возможные осложнения
Липоидоз возникает с развитием недостаточности жизненно важных органов. Наиболее распространенными осложнениями являются заболевания сердца и сосудов, центральной нервной системы, почек, легких и печени. Пациенты страдают от атеросклероза сосудов, сердечной и дыхательной недостаточности, ишемической болезни сердца, хронической почечной и печеночной недостаточности, аденом и циррозов, инсультов и преходящих ишемических атак. Для типов болезней, которые совместимы с жизнью, двигательное и умственное развитие часто нарушается. Многие пациенты не могут позаботиться о себе, обучить и освоить профессию и требуют пожизненного ухода от других.
Диагностика
Липоидозы не являются узкоспециализированными заболеваниями, поэтому в их выявлении и лечении участвуют педиатры, гематологи, гастроэнтерологи, ревматологи, неврологи, психиатры и генетики. На начальном этапе диагностики собирается семейный анамнез: при наследственной природе ферментопатии у пациента могут быть родственники с подтвержденным диагнозом липидоз. При сборе клинических данных специалисты обращают внимание на момент появления симптомов: большую часть времени заболевание проявляется в периоде новорожденных или на первом году жизни, реже у детей старшего возраста или взрослых. Конкретные методы обследования пациентов включают в себя:
• Анализ дефектной активности фермента. Рассматриваются различные биоматериалы - плазма крови, лейкоциты, пятна высохшей крови, культура фибробластов кожи, материал биопсии почки, печень. При липоидозе определяется отсутствие активности специфического фермента: от незначительного снижения до полного отсутствия.
• Количественное исследование липидов. В крови и биопсийном материале органов анализируется содержание патологически накопленных липидов и промежуточных продуктов их метаболизма. У пациентов с липоидозом показатели превышают норму. Параллельно изучаются структурные изменения в пораженных клетках.
• Секвенирование ДНК. Обнаружение дефектного гена в хромосоме является наиболее точным, но трудоемким методом диагностики наследственных заболеваний. Он широко используется в рамках перинатальной и предимплантационной диагностики, в тех случаях, когда вышеуказанные тесты не дают однозначного представления о диагнозе.
• Исследования изображений органов. Дополнительно проводится диагностика состояния пораженных органов - сердца, печени, желчного пузыря, селезенки, легких, почек и головного мозга. Использовали УЗИ, МРТ, КТ, ЭКГ, ЭЭГ. Процедуры позволяют оценить размеры, выявить структурные изменения и новообразования в организме.
• Анализ дефектной активности фермента. Рассматриваются различные биоматериалы - плазма крови, лейкоциты, пятна высохшей крови, культура фибробластов кожи, материал биопсии почки, печень. При липоидозе определяется отсутствие активности специфического фермента: от незначительного снижения до полного отсутствия.
• Количественное исследование липидов. В крови и биопсийном материале органов анализируется содержание патологически накопленных липидов и промежуточных продуктов их метаболизма. У пациентов с липоидозом показатели превышают норму. Параллельно изучаются структурные изменения в пораженных клетках.
• Секвенирование ДНК. Обнаружение дефектного гена в хромосоме является наиболее точным, но трудоемким методом диагностики наследственных заболеваний. Он широко используется в рамках перинатальной и предимплантационной диагностики, в тех случаях, когда вышеуказанные тесты не дают однозначного представления о диагнозе.
• Исследования изображений органов. Дополнительно проводится диагностика состояния пораженных органов - сердца, печени, желчного пузыря, селезенки, легких, почек и головного мозга. Использовали УЗИ, МРТ, КТ, ЭКГ, ЭЭГ. Процедуры позволяют оценить размеры, выявить структурные изменения и новообразования в организме.
Лечение
Терапия этой группы заболеваний является сложной задачей для врачей разных специальностей. Методы лечения несовершенны и продолжают развиваться, при некоторых типах заболеваний можно добиться лишь временного улучшения самочувствия пациента, в то время как в других достижима стабильная ремиссия. Общая схема оказания медицинской помощи больным состоит из трех компонентов:
• Ферменто-заместительная терапия. Препараты с искусственно изолированным дефицитным ферментом вводятся в организм пациента. Инъекции выполняются на всю жизнь, позволяют восстановить липидный обмен.
• Субстрат-редуцирующая терапия. Лечение направлено на снижение интенсивности образования патологически накапливаемых соединений (сложный липид, его метаболиты). Используются низкомолекулярные молекулы, которые стимулируют остаточную активность фермента.
• Симптоматическая терапия. Лекарственные средства подбираются индивидуально исходя из клинической картины заболевания. Распространено применение противосудорожных, анальгетиков, ингибиторов АПФ, гепатопротекторов. При некоторых типах липоидоза использование симптоматических лекарств является единственным способом лечения.
• Ферменто-заместительная терапия. Препараты с искусственно изолированным дефицитным ферментом вводятся в организм пациента. Инъекции выполняются на всю жизнь, позволяют восстановить липидный обмен.
• Субстрат-редуцирующая терапия. Лечение направлено на снижение интенсивности образования патологически накапливаемых соединений (сложный липид, его метаболиты). Используются низкомолекулярные молекулы, которые стимулируют остаточную активность фермента.
• Симптоматическая терапия. Лекарственные средства подбираются индивидуально исходя из клинической картины заболевания. Распространено применение противосудорожных, анальгетиков, ингибиторов АПФ, гепатопротекторов. При некоторых типах липоидоза использование симптоматических лекарств является единственным способом лечения.
Список литературы
1. Лизосомные болезни накопления липидов у детей/ Захарова И.Н. Медицинский совет. 2016 - №1.
2. Клиническая генетика/ Давиденкова Е.Ф., Либерман И.С. 1975.
3. Наследственные нарушения обмена липидов: учебно-методическое пособие/ Якимович Н.И. 2016.
2. Клиническая генетика/ Давиденкова Е.Ф., Либерман И.С. 1975.
3. Наследственные нарушения обмена липидов: учебно-методическое пособие/ Якимович Н.И. 2016.
Связанные клинические рекомендации
Связанные стандарты мед. помощи
- Стандарт медицинской помощи взрослым при болезни Фабри
- Стандарт медицинской помощи взрослым при болезни Ниманна-Пика тип С
- Стандарт медицинской помощи больным с другими нарушениями накопления липидов (болезнью Гоше)
- Стандарт медицинской помощи детям при болезни Ниманна-Пика, тип C
- Стандарт первичной медико-санитарной помощи детям при других нарушениях накопления липидов (болезни Гоше)