Другие названия и синонимы
Andersen syndrome, Синдром Андерсена-Тавила.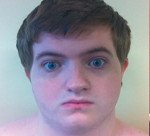
МКБ-10 коды
|
|
Описание
Редкое наследственное состояние, характеризующееся удлиненным интервалом QT и U-волнами высокой амплитуды на электрокардиограмме, желудочковой аритмией, кризами мышечного паралича и наличием внешних признаков дисморфогенеза. У пациентов определяются низкие уши, широкий лоб, аномально увеличенная челюсть, стабильная деформация пальцев с сращением и перепонками, большее расстояние между глазами, низкий рост и сколиоз. Диагноз устанавливается на основании клинических данных, результатов генетического исследования и электрокардиограммы. Симптоматическое лечение включает прием антагонистов кальция и бета-блокаторов, ограничение физической активности.
Дополнительные факты
В 1971 году датский врач Э. Андерсен описал симптомы дисморфогенеза, желудочковых аритмий и приступов паралича у 8-летнего мальчика. В 1994 году невролог Р. Тавиль более тщательно изучил клиническую картину заболевания и отличил временный паралич при этом заболевании от других форм периодического паралича. В своей работе он впервые использовал термин «синдром Андерсена». В настоящее время используется другое название - синдром Андерсена-Тавила. Эпидемиология крайне низкая: один случай заболевания диагностируется на 1 миллион новорожденных. Заболеваемость одинакова для девочек и мальчиков, а также для разных рас и этнических групп. Синдром Андерсена следует отличать от болезни Андерсена - наследственного нарушения накопления гликогена в органах и тканях.
Причины
В 2001 году была определена генетическая основа синдрома Андерсена: изучая клинические случаи в семье из 15 человек, исследователи обнаружили связь между наследованием заболевания и наличием гетерозиготных мутаций в гене KCNJ2, расположенном в локусе 23 17q хромосомы. Этот ген отвечает за синтез альфа-субъединицы белка Kir2.1 в поступающих калиевых каналах сердечной и скелетной мышц.
Наследственная передача синдрома происходит по аутосомно-доминантному механизму, и были выявлены единичные спорадические случаи - спонтанная первичная мутация мутации при зачатии. В случае аутосомно-доминантного наследования вероятность рождения больного ребенка в паре, в которой один из родителей имеет дефектный ген, составляет 50%. Проникновение заболевания очень изменчиво, поэтому не у всех пациентов имеются обширные симптомы. Тяжесть патологии варьируется в пределах семьи: у некоторых пациентов она проявляется выраженными кардиологическими и миопатическими нарушениями, у других - бессимптомно.
Наследственная передача синдрома происходит по аутосомно-доминантному механизму, и были выявлены единичные спорадические случаи - спонтанная первичная мутация мутации при зачатии. В случае аутосомно-доминантного наследования вероятность рождения больного ребенка в паре, в которой один из родителей имеет дефектный ген, составляет 50%. Проникновение заболевания очень изменчиво, поэтому не у всех пациентов имеются обширные симптомы. Тяжесть патологии варьируется в пределах семьи: у некоторых пациентов она проявляется выраженными кардиологическими и миопатическими нарушениями, у других - бессимптомно.
Патогенез
Патогенетическая основа синдрома Андерсена - отсутствие продукции белковых молекул Kir2.1. Они попадают в структуру калиевых каналов клеток, способных к возбудимости, - кардиомиоцитов, нейронов мозга и поперечно-полосатых мышечных клеток. Калиевые каналы регулируют потенциал покоя мембраны скелета и сердечной мышцы. С их дисфункцией возбудимость ткани изменяется патологически: выход ионов K + из клеток с гиперполяризованной мембраной нарушается в последней фазе возможной реполяризации.
Транскрипция гена KCNJ2, который содержит большое количество Kir2.1, обнаруживается в тканях сердца (особенно в предсердиях и желудочках), в мозге, в плаценте, в скелетных мышцах и в легких. Α-субъединицы Kir2.1 образуют тетрамер внутри клеточных стенок и образуют калиевый канал. При синдроме Андерсена синтезируется недостаточное количество этих субъединиц. В результате происходит деполяризация и дестабилизация потенциала покоя мембраны покоя, желудочковых аритмий, сокращений мышц и паралича.
Транскрипция гена KCNJ2, который содержит большое количество Kir2.1, обнаруживается в тканях сердца (особенно в предсердиях и желудочках), в мозге, в плаценте, в скелетных мышцах и в легких. Α-субъединицы Kir2.1 образуют тетрамер внутри клеточных стенок и образуют калиевый канал. При синдроме Андерсена синтезируется недостаточное количество этих субъединиц. В результате происходит деполяризация и дестабилизация потенциала покоя мембраны покоя, желудочковых аритмий, сокращений мышц и паралича.
Клиническая картина
Полная клиническая картина синдрома Андерсена представлена триадой симптомов, включая периодический калий-зависимый паралич, нарушения желудочкового ритма, нарушения в структуре черепа и конечностей. При легких формах заболевания переходный мышечный паралич ощущается как кратковременные приступы слабости в ногах и руках. Умеренное и тяжелое течение синдрома сопровождается полным или частичным параличом конечностей, генерализованным парезом с потерей чувствительности и возможностью произвольных движений. Эти эпизоды обычно происходят после интенсивной физической активности или утреннего пробуждения, внезапно в состоянии расслабления и отдыха. Пусковые факторы паралича включают голодание пешком, обильное потребление пищи, физическую активность после сидения, простуду, вдыхание токсичных газов.
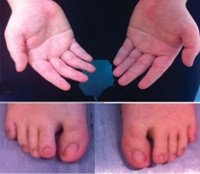
Аритмии являются наиболее распространенным симптомом заболевания. У некоторых пациентов они выявляются только по ЭКГ. Субъективно может возникнуть ощущение сердцебиения и аритмии, учащения сердцебиения, дискомфорта в груди и боли, головокружение, потеря сознания и ощущения, близкие к обмороку. Признаки дисморфогенеза представлены коротким ростом; макроцефалия - увеличение размеров черепа на 10% и более от возрастной нормы; долихоцефалия - удлинение черепа в переднезаднем направлении из-за преждевременного окостенения протекающих швов. Также наблюдается гипертелоризм - широкая система парных органов, например глаз. Уши опущены и повернуты назад. У некоторых пациентов развивается эпикант - складка во внутреннем углу глаза. Твердое и мягкое небо часто остается закрытым, верхняя челюсть небольшая (микрогнатия), пальцы неестественно укорочены (брахидактилия), пятые пальцы согнуты вбок и медиально (клинодактилия). Иногда наблюдаются суставы пальцев и перепонки.
Ассоциированные симптомы: Боль в грудной клетке. Деформация пальцев ног. Деформация пальцев рук. Слабость мышц (парез).
Аритмии являются наиболее распространенным симптомом заболевания. У некоторых пациентов они выявляются только по ЭКГ. Субъективно может возникнуть ощущение сердцебиения и аритмии, учащения сердцебиения, дискомфорта в груди и боли, головокружение, потеря сознания и ощущения, близкие к обмороку. Признаки дисморфогенеза представлены коротким ростом; макроцефалия - увеличение размеров черепа на 10% и более от возрастной нормы; долихоцефалия - удлинение черепа в переднезаднем направлении из-за преждевременного окостенения протекающих швов. Также наблюдается гипертелоризм - широкая система парных органов, например глаз. Уши опущены и повернуты назад. У некоторых пациентов развивается эпикант - складка во внутреннем углу глаза. Твердое и мягкое небо часто остается закрытым, верхняя челюсть небольшая (микрогнатия), пальцы неестественно укорочены (брахидактилия), пятые пальцы согнуты вбок и медиально (клинодактилия). Иногда наблюдаются суставы пальцев и перепонки.
Ассоциированные симптомы: Боль в грудной клетке. Деформация пальцев ног. Деформация пальцев рук. Слабость мышц (парез).
Возможные осложнения
Развитие опасных для жизни состояний возможно у пациентов с тяжелыми формами заболевания, особенно если нет необходимого лечения и ухода. Частые приступы паралича сопровождаются риском травм: пациенты внезапно теряют контроль над мышечным тонусом, падают, получают синяки и травмы. Такие условия особенно опасны при движении, пересечении улицы При отсутствии терапевтического контроля желудочковой аритмии существует небольшая вероятность внезапной смерти из-за остановки сердца. В научной литературе описано несколько случаев подобных смертей.
Диагностика
Признаки патологии можно обнаружить уже на первом году жизни ребенка. Педиатры отмечают дефекты формирования пальцев рук и ног, черепа и особенно его передней части. При легком варианте заболевания внешние симптомы слабо выражены или отсутствуют, а затем диагноз ставится позже, после случайного обнаружения изменений в ритме сердца. Обследование пациента проводят кардиологи, неврологи, генетики. Дифференциальная диагностика направлена на устранение других видов аритмий. Следующие процедуры подтверждают наличие синдрома Андерсена:
• Клинические и анамнестические исследования. Собран семейный анамнез, проведен генеалогический анализ. У большинства пациентов есть близкие родственники с подтвержденным диагнозом синдрома. При осмотре характерны черепно-лицевые аномалии, изменения структуры пальцев. Во время обследования могут предъявляться жалобы на мышечную слабость или паралич, дискомфорт в груди и неравномерное сердцебиение.
• ЭКГ. По данным электрокардиографии определяется удлинение интервала QT: в среднем у женщин - 479 мс, у мужчин - 493 мс. Другим диагностическим критерием является высокая амплитуда U-волны, которая в основном регистрируется в передних отведениях грудной клетки. Расширенные и двухфазные U-образные зубцы, нисходящая часть Т-зубцов вытянута, T-U-слияние широкое. Эпизоды обморока сопровождаются пируэтами желудочковой тахикардии.
• Генетическое тестирование. Анализ крови на вену проводится. Исследуется структура гена, ответственного за развитие заболевания (KCNJ2). Примерно у 70-80% пациентов обнаруживаются мутации в гетерозиготных и гетерозиготных сопутствующих состояниях. Тип мутации индивидуален. Наиболее распространенной гетерозиготной миссенс-мутацией является R218W.
• Клинические и анамнестические исследования. Собран семейный анамнез, проведен генеалогический анализ. У большинства пациентов есть близкие родственники с подтвержденным диагнозом синдрома. При осмотре характерны черепно-лицевые аномалии, изменения структуры пальцев. Во время обследования могут предъявляться жалобы на мышечную слабость или паралич, дискомфорт в груди и неравномерное сердцебиение.
• ЭКГ. По данным электрокардиографии определяется удлинение интервала QT: в среднем у женщин - 479 мс, у мужчин - 493 мс. Другим диагностическим критерием является высокая амплитуда U-волны, которая в основном регистрируется в передних отведениях грудной клетки. Расширенные и двухфазные U-образные зубцы, нисходящая часть Т-зубцов вытянута, T-U-слияние широкое. Эпизоды обморока сопровождаются пируэтами желудочковой тахикардии.
• Генетическое тестирование. Анализ крови на вену проводится. Исследуется структура гена, ответственного за развитие заболевания (KCNJ2). Примерно у 70-80% пациентов обнаруживаются мутации в гетерозиготных и гетерозиготных сопутствующих состояниях. Тип мутации индивидуален. Наиболее распространенной гетерозиготной миссенс-мутацией является R218W.
|
|
Лечение
Этиотронное лечение не разработано; попытки использовать высокие дозы препаратов калия неэффективны. Основным методом терапии является назначение бета-адреноблокаторов и блокаторов кальциевых каналов (фенилалкиламин). Эти препараты уменьшают симптомы аритмии, снижают силу и частоту сердечных сокращений, подавляют сердечную проводимость. В тяжелых случаях синдрома обморока проводится имплантация кардиовертера-дефибриллятора. Всем пациентам рекомендуется корректировать свой образ жизни: запрещено принимать препараты, продлевающие интервал QT (антиангинальные, антидиуретические, липидоснижающие, психотропные и некоторые другие препараты), необходимо ограничивать физическую активность и другие факторы, провоцирующие приступы паралича.
Список литературы
1. Синдром Андерсена/ Крупянко С.М., Какучая Т.Т. Анналы аритмологии. 2005.
2. Редкий вариант первичного синдрома удлиненного интервала QT - синдром Андерсена-Тавила/ Соловьев В.М., Подшивалова О.Ю., Ковалев И.А., Ильдарова Р.А., Школьникова М.А. Педиатрия. Журнал им. Г.Н. Сперанского. 2015.
3. Синдром Ангельмана (этиология и патогенез)/ Абатуров А.Е., Петренко Л.Л., Кривуша Е.Л. Здоровье ребенка. 2015.
2. Редкий вариант первичного синдрома удлиненного интервала QT - синдром Андерсена-Тавила/ Соловьев В.М., Подшивалова О.Ю., Ковалев И.А., Ильдарова Р.А., Школьникова М.А. Педиатрия. Журнал им. Г.Н. Сперанского. 2015.
3. Синдром Ангельмана (этиология и патогенез)/ Абатуров А.Е., Петренко Л.Л., Кривуша Е.Л. Здоровье ребенка. 2015.