Другие названия и синонимы
Olivopontocerebellar degeneration.
МКБ-10 коды
|
|
Описание
Оливопонтоцеребеллярные дегенерации. Наследственные дегенеративные заболевания ЦНС, объединенные сходной локализацией патологического процесса в мозжечке, нижних оливах и мосте головного мозга. Клиника складывается из мозжечкового синдрома, экстрапирамидных расстройств, когнитивных и психических нарушений. Диагностируются оливопонтоцеребеллярные дегенерации на основании анамнеза, генеалогического исследования, данных неврологического и психологического обследования, результатов КТ и МРТ головного мозга. Терапия симптоматическая, включает нейропротекторные и общеукрепляющие фармпрепараты, ЛФК, массаж. Прогноз неблагоприятный.
Дополнительные факты
Прогрессирующий мозжечковый синдром в сочетании с экстрапирамидными нарушениями и психическими расстройствами был описан Дежерином и Томасом в 1900 году. В последующем были выделены несколько форм данной патологии, объединенные в единую группу заболеваний, получившую название «оливопонтоцеребеллярные дегенерации» (ОПЦД). По сути, речь идет о заболеваниях с единой локализацией мультифокальной дегенерации и гибели нейронов. Именно преимущественное расположение дегенеративных процессов легло в основу названия этой группы поражений ЦНС (с латинского oliva - олива, pont(is) - мост, cerebellum - мозжечок). В ряде случаев наблюдается поражение каудальных черепно-мозговых нервов (IX, X, XI, XII пар), реже - передних рогов спинного мозга и проводящих трактов.
Оливопонтоцеребеллярные дегенерации входят в группу дегенеративных поражений ЦНС, к которой относятся болезнь Паркинсона, рассеянный склероз, болезнь Альцгеймера, лейкодистрофии, болезнь Пика, спинальные амиотрофии и мн. тд Отмечается аутосомное рецессивное и доминантное наследование, спорадические случаи. Возраст манифестации клинических проявлений варьирует в пределах от 11 до 80 лет, наиболее часто дебют происходит в четверной или пятой декаде жизни.
Оливопонтоцеребеллярные дегенерации входят в группу дегенеративных поражений ЦНС, к которой относятся болезнь Паркинсона, рассеянный склероз, болезнь Альцгеймера, лейкодистрофии, болезнь Пика, спинальные амиотрофии и мн. тд Отмечается аутосомное рецессивное и доминантное наследование, спорадические случаи. Возраст манифестации клинических проявлений варьирует в пределах от 11 до 80 лет, наиболее часто дебют происходит в четверной или пятой декаде жизни.
Причины
Точные представления о этиопатогенезе ОПЦД пока не сформированы. Поиск генетического субстрата дегенераций привел к выявлению нарушений в локусе 6p22-23 (при дегенерации Менделя) и в локусе 12q23-24 (при дегенерации Фиклера-Винклера) в виде увеличения числа тринуклеотидных повторов. У ряда пациентов наблюдается недостаточность дегидрогеназы глутаминовой кислоты, необходимой для метаболизма глутамата. Последний в качестве медиатора активирует передачу возбуждения от мозжечковой коры к клеткам Пуркинье, аксоны которых формируют эфферентные (нисходящие) мозжечковые тракты. Избыточное накопление глутамата при дефиците дегидрогеназы оказывает нейротоксический эффект, который, возможно, является основной причиной дегенеративных изменений клеток Пуркинье.
Основными морфологическими признаками ОПЦД выступают: асимметричные атрофические изменения белого вещества мозжечковых полушарий и в меньшей степени его червя, дегенерация средней и нижней мозжечковых ножек, глиоз и сморщивание ядер моста и олив. Атрофия коры мозжечка с утратой клеток Пуркинье отмечается на более поздних стадиях ОПЦД. Типична полная интактность верхней ножки, узелка червя (nodulus) и клочка (flocculus) мозжечка. Гистологический анализ пораженных церебральных тканей выявляет дистрофически-дегенеративные изменения нейронов, разрастание глии, демиелинизацию нервных волокон.
Основными морфологическими признаками ОПЦД выступают: асимметричные атрофические изменения белого вещества мозжечковых полушарий и в меньшей степени его червя, дегенерация средней и нижней мозжечковых ножек, глиоз и сморщивание ядер моста и олив. Атрофия коры мозжечка с утратой клеток Пуркинье отмечается на более поздних стадиях ОПЦД. Типична полная интактность верхней ножки, узелка червя (nodulus) и клочка (flocculus) мозжечка. Гистологический анализ пораженных церебральных тканей выявляет дистрофически-дегенеративные изменения нейронов, разрастание глии, демиелинизацию нервных волокон.
Классификация
В настоящее время в клинической неврологии известны 5 основных типов оливопонтоцеребеллярных дегенераций. Отдельно выделяют синдром Шая-Дрейджера, который наряду с оливопонтоцеребеллярной дегенерацией включает диффузную церебральную атрофию и дегенерацию стрионигральных структур.
Tип I Менделя. Аутосомно - доминантная ОПЦД с дебютом после 11 лет и до 60 - летнего возраста. Характерна мозжечковая атаксия, гиперкинезы, гипотония мышц, дисфагия. Реже встречаются пирамидные расстройства (парезы конечностей, гипестезия).
Tип II Фиклера. Винклера - аутосомно - рецессивная ОПЦД, манифестирующая с третьей декады жизни до 80 - летнего возраста. Протекает без нарушений чувствительности и парезов. Глубокие рефлексы сохранены.
Tип III ОПЦД с ретинальной дегенерацией. Аутосомно - доминантная форма, поражающая преимущественно лиц молодого возраста. Характеризуется мозжечковым синдромом и гиперкинезами в сочетании с прогрессирующим падением зрения вследствие пигментной ретинопатии.
Tип IV Шута. Хайкмана - аутосомно - доминантная ОПЦД детского и молодого возраста. Типичные для всех ОПЦД мозжечковые расстройства сочетаются с поражением каудальных черепных нервов (бульбарные симптомы) и задних столбов спинного мозга (нарушение глубокой чувствительности).
Tип V ОПЦД с деменцией, экстрапирамидными знаками и офтальмоплегией. Наследуется аутосомно-доминантно. Представляет собой комбинацию указанных в названии синдромов и мозжечковой атаксии.
Tип I Менделя. Аутосомно - доминантная ОПЦД с дебютом после 11 лет и до 60 - летнего возраста. Характерна мозжечковая атаксия, гиперкинезы, гипотония мышц, дисфагия. Реже встречаются пирамидные расстройства (парезы конечностей, гипестезия).
Tип II Фиклера. Винклера - аутосомно - рецессивная ОПЦД, манифестирующая с третьей декады жизни до 80 - летнего возраста. Протекает без нарушений чувствительности и парезов. Глубокие рефлексы сохранены.
Tип III ОПЦД с ретинальной дегенерацией. Аутосомно - доминантная форма, поражающая преимущественно лиц молодого возраста. Характеризуется мозжечковым синдромом и гиперкинезами в сочетании с прогрессирующим падением зрения вследствие пигментной ретинопатии.
Tип IV Шута. Хайкмана - аутосомно - доминантная ОПЦД детского и молодого возраста. Типичные для всех ОПЦД мозжечковые расстройства сочетаются с поражением каудальных черепных нервов (бульбарные симптомы) и задних столбов спинного мозга (нарушение глубокой чувствительности).
Tип V ОПЦД с деменцией, экстрапирамидными знаками и офтальмоплегией. Наследуется аутосомно-доминантно. Представляет собой комбинацию указанных в названии синдромов и мозжечковой атаксии.
Клиническая картина
Базисным клиническим проявлением ОПЦД является мозжечковая атаксия. Дебют заболевания знаменуется появлением легкой неустойчивости, неуклюжести при беге и быстрой ходьбе. Прогрессирование этих симптомов приводит к выраженным расстройствам походки и статики. Ходьба затруднена, сопровождается падениями, во избежание которых пациенты широко расставляют ноги во время ходьбы. Грубые нарушения равновесия обуславливают колебания туловища пациента, когда он стоит или сидит. Позднее присоединяется дискоординация в конечностях: адиадохокинез, гипер- и дисметрия, крупноразмашистый почерк. Атаксия в конечностях сопровождается дрожанием головы и интенционным тремором. Наблюдается горизонтальный нистагм. Одновременно с атаксией в конечностях появляется типичная мозжечковая дизартрия, т. н. «скандированная речь». Как правило, происходит повышение рефлексов, в отдельных случаях - их понижение.
Возможны расстройства глотания (дисфагия), гиперкинезы, симптомы вторичного паркинсонизма, лицевой парез, пирамидная недостаточность. При поражении ядер каудальных черепных нервов возникает офтальмоплегия, бульбарный паралич. В большинстве случаев оливопонтоцеребеллярные дегенерации протекают с недержанием мочи. В эмоциональной сфере преобладает снижение: вялость, безынициативность, тупость. Обычно отмечаются значительные когнитивные нарушения и деменция. Характерны психические расстройства: галлюцинаторный синдром, депрессия, фобические расстройства, эпизоды психомоторного возбуждения, спутанность сознания. Иногда их появление предшествует развитию мозжечковой атаксии.
Ассоциированные симптомы: Общая слабость. Галлюцинации. Замедленность движений (брадикинезия). Скандированная речь. Тремор. Тремор головы. Тремор при движении. Шаркающая походка. Шаткая походка.
Возможны расстройства глотания (дисфагия), гиперкинезы, симптомы вторичного паркинсонизма, лицевой парез, пирамидная недостаточность. При поражении ядер каудальных черепных нервов возникает офтальмоплегия, бульбарный паралич. В большинстве случаев оливопонтоцеребеллярные дегенерации протекают с недержанием мочи. В эмоциональной сфере преобладает снижение: вялость, безынициативность, тупость. Обычно отмечаются значительные когнитивные нарушения и деменция. Характерны психические расстройства: галлюцинаторный синдром, депрессия, фобические расстройства, эпизоды психомоторного возбуждения, спутанность сознания. Иногда их появление предшествует развитию мозжечковой атаксии.
Ассоциированные симптомы: Общая слабость. Галлюцинации. Замедленность движений (брадикинезия). Скандированная речь. Тремор. Тремор головы. Тремор при движении. Шаркающая походка. Шаткая походка.
Диагностика
Постановка диагноза требует сопоставления времени и симптомов дебюта заболевания, данных неврологического статуса (сочетание мозжечковых нарушений с гиперкинезами) и нейропсихологического обследования (наличие когнитивного снижения, отклонения в эмоциональной сфере) с результатами нейровизуализации.
|
|
Дифференциальная диагностика
Дифференцировать оливопонтоцеребеллярные дегенерации необходимо от атаксии Пьера-Мари и атаксии Фридрейха, опухолей мозжечка, прогрессирующих вариантов рассеянного склероза, дисметаболических заболеваний с мозжечковым синдромом (например, болезни Рефсума).
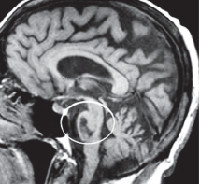
Компьютерная томография малоинформативна, поскольку определяет преимущественно неспецифические изменения церебральных структур: расширение желудочков и субарахноидальных пространств. Специфичным признаком, регистрируемым при помощи КТ головного мозга, является уменьшение толщины передней мозжечковой ножки. Более полно диагностировать оливопонтоцеребеллярные дегенерации позволяет МРТ головного мозга. С ее помощью можно визуализировать атрофические изменения в мосте и продолговатом мозге. Для определения типа наследования патологии необходима консультация генетика и генеалогическое исследование. При подозрении на ОПЦД I или II типов возможна ДНК-диагностика. Пациенты с нарушением зрения нуждаются в консультации офтальмолога.
Компьютерная томография малоинформативна, поскольку определяет преимущественно неспецифические изменения церебральных структур: расширение желудочков и субарахноидальных пространств. Специфичным признаком, регистрируемым при помощи КТ головного мозга, является уменьшение толщины передней мозжечковой ножки. Более полно диагностировать оливопонтоцеребеллярные дегенерации позволяет МРТ головного мозга. С ее помощью можно визуализировать атрофические изменения в мосте и продолговатом мозге. Для определения типа наследования патологии необходима консультация генетика и генеалогическое исследование. При подозрении на ОПЦД I или II типов возможна ДНК-диагностика. Пациенты с нарушением зрения нуждаются в консультации офтальмолога.
Лечение
Специфическая терапия ОПЦД пока не найдена, поэтому неврологами осуществляется симптоматическое лечение, направленное на уменьшение конкретных клинических проявлений. Показаны нейрометаболиты и общеукрепляющие средства: витамины гр. В, витамин С и тд Для нормализации мышечного тонуса проводится массаж и ЛФК. С целью уменьшения выраженности нарушений координации рекомендованы специальные упражнения для ее тренировки. При синдроме паркинсонизма показаны центральные холинолитики: диэтазина гидрохлорид, тригексифенидил.
Прогноз
Несмотря на проводимое лечение, оливопонтоцеребеллярные дегенерации имеют неуклонно прогрессирующее течение. Длительность заболевания в среднем колеблется в пределах 10-15 лет, иногда достигает 20 лет. Причиной летального исхода, как правило, являются интеркуррентные инфекции (застойная пневмония, сепсис).