Другие названия и синонимы
Machado-Joseph Disease, Спиноцеребеллярная атаксия III тип, СЦА З тип.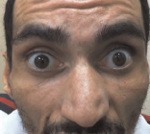
МКБ-10 коды
|
|
Описание
Болезнь Мачадо. Джозефа - генетически обусловленная спиноцеребеллярная атаксия, клинически представленная полиморфными сочетаниями мозжечкового синдрома с проявлениями вторичного паркинсонизма, гиперкинезами, пирамидными расстройствами в виде спастических параличей и офтальмоплегии, амиотрофиями. Диагностируется на основании тщательного изучения клинических проявлений у пациента и его родственников, генеалогического анализа, данных МРТ и КТ, выявления при исследовании ДНК превышающего норму количества копий триплета ЦАГ. Лечение симптоматическое. Прогноз неблагоприятный.
Дополнительные факты
Болезнь Мачадо-Джозефа описана в средине 70-х годов 20 века. Предположительно, заболевание первоначально возникло у жителей Азорских островов, в связи с чем оно иногда встречается под названием «азорская болезнь». На сегодняшний день болезнь Мачадо-Джозефа распространена по всему миру. Ее случаи диагностированы у жителей США, Бразилии, Японии, Индии, Европы, Китая, Австралии, Канады. Она является наиболее встречаемой формой наследственной мозжечковой атаксии.
В рамках современной международной классификации заболеваний данная патология верифицируется как СЦА З - спиноцеребеллярная атаксия III тип. Характерна большая вариабельность времени дебюта (от 10 до 70 лет) и полиморфность клинических симптомов, обусловленная мультисистемным поражением церебральных и спинальных структур. В зависимости от комбинации основных клинических синдромов различают 3 варианта заболевания. Полиморфизм проявлений влечет за собой определенные затруднения в диагностике заболевания, которая может быть правильно реализована только при тесном сотрудничестве специалистов в области неврологии и генетики.
В рамках современной международной классификации заболеваний данная патология верифицируется как СЦА З - спиноцеребеллярная атаксия III тип. Характерна большая вариабельность времени дебюта (от 10 до 70 лет) и полиморфность клинических симптомов, обусловленная мультисистемным поражением церебральных и спинальных структур. В зависимости от комбинации основных клинических синдромов различают 3 варианта заболевания. Полиморфизм проявлений влечет за собой определенные затруднения в диагностике заболевания, которая может быть правильно реализована только при тесном сотрудничестве специалистов в области неврологии и генетики.
Клиническая картина
Ассоциированные симптомы: Парапарез. Тремор.
Причины
Ранее этиология была неизвестна. Благодаря развитию ДНК-исследований, выяснилось, что основным субстратом патологии выступает генная мутация, которая передается потомству аутосомно-доминантным путем. Аберрация локализуется в 14-й хромосоме (локус 14q24. 3-q32) и заключается в экспансии (увеличении количества повторов) тринуклеотидного сочетания «цитозин-аденин-гуанин». Число повторов триплета ЦАГ существенно варьирует и в среднем составляет 62-84, в то время как в норме не превышает 37. Чем оно больше, тем ранее возникает манифестация болезни.
Морфологически наблюдается апоптоз нейронов зернистого слоя и клеток Пуркинье в коре мозжечка, дегенеративные изменения зубчатого и красного ядер, черной субстанции, моторных ядер черепно-мозговых нервов и передних рогов спинного мозга, спиномозжечковых трактов. В полосатом теле выявляются глиальные разрастания. Отличительной особенностью является интактность олив продолговатого мозга.
Морфологически наблюдается апоптоз нейронов зернистого слоя и клеток Пуркинье в коре мозжечка, дегенеративные изменения зубчатого и красного ядер, черной субстанции, моторных ядер черепно-мозговых нервов и передних рогов спинного мозга, спиномозжечковых трактов. В полосатом теле выявляются глиальные разрастания. Отличительной особенностью является интактность олив продолговатого мозга.
Классификация
Болезнь Мачадо. Джозефа I тип манифестирует в возрастном периоде 10 - 30 лет. Характеризуется сочетанием пирамидных и экстрапирамидных симптомов. Пирамидный синдром (поражение кортикоспинальных трактов) обычно дебютирует спастическим парапарезом, затем присоединяется слабость в руках, парез мышц глотки с развитием дисфагии и дизартрии, парез глазодвигательных нервов с офтальмоплегией (симптом «фиксированных глазных яблок»). Наблюдается клонус стоп, патологические рефлексы. Экстрапирамидный синдром проявляется симптомами торсионной дистонии, атетозом, вторичным паркинсонизмом. Формируется скованная медленная походка с широкой расстановкой ног. Отмечается шаткость при ходьбе, обусловленная спастикой мышц, а не атаксией. Типичен экзофтальм, крупные фасцикулярные сокращения языка, не сопровождающиеся его атрофией. Возможны фасцикуляции мимических мышц, миокимия век. Наблюдается вертикальный и горизонтальный нистагм, саккады (однонаправленные движения глаз) с повышенной/пониженной амплитудой.
Болезнь Мачадо. Джозефа II тип дебютирует в период от 20 до 40 лет. Проявляется симптомами мозжечковой атаксии: абазией, гипер- и дисметрией, нарушением равновесия, дизартрией. Типично сочетание мозжечковой симптоматики с пирамидными и экстрапирамидными проявлениями, встречающимися при типе I. Офтальмоплегия и фасцикуляции наблюдаются гораздо реже, чем при болезни Мачадо-Джозефа I типа.
Болезнь Мачадо. Джозефа III тип представляет собой комбинацию мозжечковой атаксии и амиотрофий. Имеет наиболее позднее начало - после 40-летнего возраста. На фоне мозжечковых симптомов наблюдаются диффузные мышечные атрофии, сопровождающиеся гипотонией и слабостью, утратой сухожильных рефлексов. Отличительной особенностью выступает наличие расстройств всех видов чувствительности по дистальному типу, свидетельствующее о полиневропатии. Экзофтальм и лицевые фасцикуляции встречаются по различным данным у 20-50% пациентов с этой формой заболевания. Дегенерация кортикоспинальных трактов и поражение экстрапирамидной системы не характерны.
Болезнь Мачадо. Джозефа II тип дебютирует в период от 20 до 40 лет. Проявляется симптомами мозжечковой атаксии: абазией, гипер- и дисметрией, нарушением равновесия, дизартрией. Типично сочетание мозжечковой симптоматики с пирамидными и экстрапирамидными проявлениями, встречающимися при типе I. Офтальмоплегия и фасцикуляции наблюдаются гораздо реже, чем при болезни Мачадо-Джозефа I типа.
Болезнь Мачадо. Джозефа III тип представляет собой комбинацию мозжечковой атаксии и амиотрофий. Имеет наиболее позднее начало - после 40-летнего возраста. На фоне мозжечковых симптомов наблюдаются диффузные мышечные атрофии, сопровождающиеся гипотонией и слабостью, утратой сухожильных рефлексов. Отличительной особенностью выступает наличие расстройств всех видов чувствительности по дистальному типу, свидетельствующее о полиневропатии. Экзофтальм и лицевые фасцикуляции встречаются по различным данным у 20-50% пациентов с этой формой заболевания. Дегенерация кортикоспинальных трактов и поражение экстрапирамидной системы не характерны.
Диагностика
Большая вариабельность количества повторов триплета ЦАГ, наблюдаемая даже в пределах одной семьи, обуславливает значительный полиморфизм клинических проявлений болезни Мачадо-Джозефа, влекущий за собой существенные диагностические трудности. Распространенным является наличие различных типов заболевания у кровных родственников, особенно, если речь идет о разных поколениях. Описаны казуистические случаи, когда паркинсонизм выступал ведущим и единственным проявлением болезни. Таким образом, ключевым моментом в диагностике болезни Мачадо-Джозефа выступает консультация генетика, подробное исследование генеалогического древа с осмотром как можно большего числа родственников больного, проведение ДНК-диагностики.
С точки зрения невролога важным является выявление специфических отличий синдрома паркинсонизма, типичных для болезни Мачадо-Джозефа. Отсутствуют патогномоничные для болезни Паркинсона постуральные расстройства и тремор покоя, а неустойчивость в положении стоя и при ходьбе связана с статико-локомоторной атаксией. Паркинсонизм оказывается устойчивым к действию препаратов леводопы, хотя в начальных стадиях болезни может наблюдаться эффект в виде уменьшения мышечной ригидности.
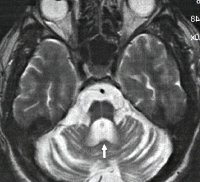
Первичная неврологическая диагностика (Эхо-ЭГ, ЭЭГ, РЭГ) не дает специфичных признаков, ее результаты могут быть в пределах нормы. КТ и МРТ головного мозга выявляет дегенерацию червя мозжечка и покрышки моста. Типичным признаком является выраженное расширение IV желудочка на фоне относительной сохранности мозжечковой коры.
С точки зрения невролога важным является выявление специфических отличий синдрома паркинсонизма, типичных для болезни Мачадо-Джозефа. Отсутствуют патогномоничные для болезни Паркинсона постуральные расстройства и тремор покоя, а неустойчивость в положении стоя и при ходьбе связана с статико-локомоторной атаксией. Паркинсонизм оказывается устойчивым к действию препаратов леводопы, хотя в начальных стадиях болезни может наблюдаться эффект в виде уменьшения мышечной ригидности.
Первичная неврологическая диагностика (Эхо-ЭГ, ЭЭГ, РЭГ) не дает специфичных признаков, ее результаты могут быть в пределах нормы. КТ и МРТ головного мозга выявляет дегенерацию червя мозжечка и покрышки моста. Типичным признаком является выраженное расширение IV желудочка на фоне относительной сохранности мозжечковой коры.
Дифференциальная диагностика
Дифференцируют болезнь Мачадо-Джозефа от других видов спиноцеребеллярных дегенерации, болезни Галлервордена-Шпатца, оливопонтоцеребеллярной дегенерации, атаксии Фридрейха, бокового амиотрофического склероза, атаксии Пьера-Мари, прогрессивного супрануклеарного паралича.
Лечение
Эффективная терапия пока не найдена. Проводится симптоматическое лечение. При синдроме паркинсонизма показаны агонисты дофамина ( прамипексол, пирибедил), может применяться амантадин. Для снятия спастики назначаются фармпрепараты с миорелаксирующим действием (толперизон, баклофен). При гиперкинезах рекомендованы производные вальпроевой к-ты или бензодиазепины (клоназепам).
|
|
Прогноз
К сожалению, во многих случаях симптоматическая терапия оказывается не способной остановить прогрессирование болезни Мачадо-Джозефа. Наблюдается неуклонное усугубление симптоматики, приводящее к гибели пациента. Продолжительность жизни после дебюта заболевания составляет от 10 до 20 лет и зависит от клинического типа патологии. Наиболее скоротечным вариантом является болезнь Мачадо-Джозефа I типа. Профилактика заключается в проведении медико-генетического консультирования и пренатальной диагностики в отягощенных семьях; недопущении рождения ребенка, имеющего соответствующую генетическую мутацию.