Другие названия и синонимы
Crouzon syndrome.
МКБ-10 коды
|
|
Описание
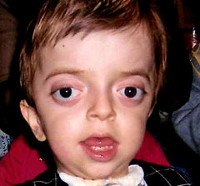
Синдром Крузона. Редкое генетическое заболевание, сопровождающееся прогрессирующими деформациями лицевой и мозговой части черепа и краниосиностозом с развитием сопутствующих нарушений. Симптомами этого состояния являются изменение формы головы (брахицефалия, скафоцефалия, тригоноцефалия), крючковидный нос, гипоплазия средней трети лица, нарушения зрения и слуха. Диагностика синдрома Крузона осуществляется на основании внешних проявлений заболевания, рентгенологических данных, а также молекулярно-генетических анализов. Специфического лечения этой патологии не существует, используются паллиативные и симптоматические мероприятия, в том числе хирургического характера.
Дополнительные факты
Синдром Крузона (краниофасциальный дизостоз 1 типа) - генетическое заболевание, характеризующееся нарушением процессов окостенения и развития элементов скелета лицевого и мозгового черепа. Впервые это состояние было описано в 1912 году французским педиатром О. Крузоном, с тех пор синдром носит его имя. Механизм наследования синдрома Крузона - аутосомно-доминантный, однако заболевание часто обусловлено спонтанными мутациями. Патология встречается достаточно редко - примерно 1,6 случаев на 100 000 новорожденных, при этом данным синдромом обусловлено почти 5% от всех пороков развития, сопровождающихся черепным дизостозом. Долгое время считалось, что это состояние имеет две разновидности - обычную и сопровождающуюся кожными нарушениями (гиперкератозом, акантозом), но с учетом современных данных специалисты в области генетики установили, что синдром Крузона с черным акантозом (CAN) является отдельным наследственным заболеванием. В то же время, патогенез его развития аналогичен классической форме заболевания, именно этим объясняется значительная схожесть симптомов. Состояние с одинаковой вероятностью поражает как мальчиков, так и девочек.
Причины
Классический вариант синдрома Крузона обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме - он кодирует аминокислотную последовательность рецептора к фактору роста фибробластов 2. Данный ген обладает значительным размером и большим количеством экзонов, что снижает его стабильность - в нем часто развиваются дефекты, приводящие к многочисленным генетическим заболеваниям, в основном поражающим элементы скелета. Так, помимо синдрома Крузона, мутации FGFR2 могут быть причиной синдромов Апера, Сетре-Чотзена, Бира-Стивенсона, синдрома Пфайффера и многих других патологий. Генетические исследования показали, что краниофасциальный дизостоз 1-го типа способны вызывать более 35 мутаций вышеуказанного гена, в основном они локализованы в области 7 и 9 экзонов.
Практически все дефекты гена FGFR2 относятся к миссенс-мутациям, то есть провоцируют изменение структуры кодируемого белка. Изменение конформации рецептора к фактору роста фибробластов 2 нарушает межклеточные взаимодействия в соединительных тканях черепа, главным образом костной и хрящевой. Это приводит сначала к накоплению фибробластов в области межкостных швов, а потом к активизации процессов окостенения, что и является причиной ведущего проявления синдрома Крузона - черепного синостоза. Некоторые исследователи полагают, что данные генетические дефекты влияют также на эмбриональное развитие структур первой жаберной дуги - к ним относят челюсти и отчасти элементы средней трети лица. Именно этим объясняется гипоплазия челюстей, особенно нижней, при синдроме Крузона.
Причины синдрома Крузона с черным акантозом несколько иные - он вызывается мутациями гена FGFR3, локализованного на 4 хромосоме. Продуктом его экспрессии также является рецептор к фактору роста фибробластов, только 3 типа (в отличие от 2 типа, являющего продуктом гена FGFR2). Выяснено, что только одна мутация этого гена выступает причиной синдрома Крузона с характерными кожными проявлениями - Ala391Glu. Это тоже миссенс-мутация, изменяющая структуру белка-рецептора. Патогенез заболевания практически не отличается от классического варианта. Изменения лица и черепа при синдроме Крузона с черным акантозом аналогичны предыдущему типу, однако к ним присоединяются гиперкератоз различных участков кожи и акантоз, нередко наблюдаются многочисленные родинки.
Практически все дефекты гена FGFR2 относятся к миссенс-мутациям, то есть провоцируют изменение структуры кодируемого белка. Изменение конформации рецептора к фактору роста фибробластов 2 нарушает межклеточные взаимодействия в соединительных тканях черепа, главным образом костной и хрящевой. Это приводит сначала к накоплению фибробластов в области межкостных швов, а потом к активизации процессов окостенения, что и является причиной ведущего проявления синдрома Крузона - черепного синостоза. Некоторые исследователи полагают, что данные генетические дефекты влияют также на эмбриональное развитие структур первой жаберной дуги - к ним относят челюсти и отчасти элементы средней трети лица. Именно этим объясняется гипоплазия челюстей, особенно нижней, при синдроме Крузона.
Причины синдрома Крузона с черным акантозом несколько иные - он вызывается мутациями гена FGFR3, локализованного на 4 хромосоме. Продуктом его экспрессии также является рецептор к фактору роста фибробластов, только 3 типа (в отличие от 2 типа, являющего продуктом гена FGFR2). Выяснено, что только одна мутация этого гена выступает причиной синдрома Крузона с характерными кожными проявлениями - Ala391Glu. Это тоже миссенс-мутация, изменяющая структуру белка-рецептора. Патогенез заболевания практически не отличается от классического варианта. Изменения лица и черепа при синдроме Крузона с черным акантозом аналогичны предыдущему типу, однако к ним присоединяются гиперкератоз различных участков кожи и акантоз, нередко наблюдаются многочисленные родинки.
Клиническая картина
Проявления синдрома Крузона можно заметить уже при рождении ребенка, однако наиболее выраженными они становятся на протяжении первых 3-4 лет жизни. Самым характерным симптомом заболевания является краниосиностоз, который может развиваться на венечном или стреловидном (намного реже) шве, прочно соединяя кости и останавливая нормальный рост головы. Сразу после рождения первые признаки синостоза могут быть стертыми, но всегда наблюдается гипертелоризм, прогнатия нижней челюсти, изменение формы носа по типу «клюва попугая», незначительный экзофтальм из-за уменьшенного размера глазниц, низкое расположение наружного слухового прохода. Иногда при синдроме Крузона выявляется синдактилия пальцев, в этом случае необходимо производить дифференциальную диагностику с синдромом Апера. У некоторых больных обнаруживается атрезия хоан, затрудняющая дыхание, а также гидроцефалия, еще больше осложняющая течение заболевания за счет резкого возрастания внутричерепного давления.
Особенностью синдрома Крузона является неминуемое прогрессирование заболевания, особенно в отношении формы черепа. Из-за образования прочного синостоза и продолжающегося роста размеров головного мозга форма головы изменяется, возникает брахицефалия или «башенный череп» - в зависимости от того, по какому шву произошло срастание. При синдроме Крузона в области сросшихся костей черепа также могут образовываться экзостозы. Такая деформация приводит и к поражению органов зрения - сначала возникает расходящееся косоглазие, затем экзофтальм сильно прогрессирует вплоть до выпадения глазных яблок из орбиты. Нередко синдром Крузона сопровождается расстройствами слуха из-за нарушения структуры пирамиды височной кости - ее полости уменьшены в размерах, некоторые из них могут отсутствовать, нередко это приводит к полной глухоте. Наблюдаются изменения и со стороны нервной системы, обнаруживаются нарастающие признаки умственной отсталости (при отсутствии паллиативных мероприятий), симптомы повышения внутричерепного давления (головные боли, рвота), судорожные припадки.
Ассоциированные симптомы: Рвота. Судороги.
Особенностью синдрома Крузона является неминуемое прогрессирование заболевания, особенно в отношении формы черепа. Из-за образования прочного синостоза и продолжающегося роста размеров головного мозга форма головы изменяется, возникает брахицефалия или «башенный череп» - в зависимости от того, по какому шву произошло срастание. При синдроме Крузона в области сросшихся костей черепа также могут образовываться экзостозы. Такая деформация приводит и к поражению органов зрения - сначала возникает расходящееся косоглазие, затем экзофтальм сильно прогрессирует вплоть до выпадения глазных яблок из орбиты. Нередко синдром Крузона сопровождается расстройствами слуха из-за нарушения структуры пирамиды височной кости - ее полости уменьшены в размерах, некоторые из них могут отсутствовать, нередко это приводит к полной глухоте. Наблюдаются изменения и со стороны нервной системы, обнаруживаются нарастающие признаки умственной отсталости (при отсутствии паллиативных мероприятий), симптомы повышения внутричерепного давления (головные боли, рвота), судорожные припадки.
Ассоциированные симптомы: Рвота. Судороги.
Диагностика
Выявление синдрома Крузона возможно на этапе пренатального развития, сразу после рождения или в первые годы жизни больного. Для этого применяются рентгенологические методики, общий осмотр, молекулярно-генетические анализы. Вспомогательную роль в диагностике синдрома Крузона играют такие методы, как офтальмологический осмотр, исследование слуха, оценка интеллектуального и психического развития. При осмотре маленьких детей определяются низко посаженные уши, гипоплазия средней трети лица, экзофтальм. У больных синдромом Крузона старшего возраста к этим проявлениям присоединяются расходящееся косоглазие, ослабление слуха вплоть до полной глухоты, изменение формы черепа. На рентгенографии черепа регистрируется синостоз в области венечного, стреловидного или лямбдовидного швов, возможно обнаружение экзостозов и уплощенной формы глазниц.
Томография пирамиды височной кости при синдроме Крузона выявляет нарушение формирования наружного слухового прохода (атрезия или стеноз) и других полостей, иногда наблюдается отсутствие барабанной полости. Турецкое седло несколько расширено, могут образовываться добавочные мелкие околоносовые синусы. Молекулярно-генетическая диагностика синдрома Крузона производится врачом-генетиком и при классической форме заболевания сводится к автоматическому секвенированию 7 и 9 экзонов гена FGFR2 с целью выявления мутаций. При наличии кожных проявлений (гиперкератоза, бородавках, множественных родинках) имеет смысл производить поиск мутации Ala391Glu в гене FGFR3. Для обеих форм синдрома Крузона возможна пренатальная генетическая диагностика, ультразвуковые методики при этом, как правило, малоэффективны.
Томография пирамиды височной кости при синдроме Крузона выявляет нарушение формирования наружного слухового прохода (атрезия или стеноз) и других полостей, иногда наблюдается отсутствие барабанной полости. Турецкое седло несколько расширено, могут образовываться добавочные мелкие околоносовые синусы. Молекулярно-генетическая диагностика синдрома Крузона производится врачом-генетиком и при классической форме заболевания сводится к автоматическому секвенированию 7 и 9 экзонов гена FGFR2 с целью выявления мутаций. При наличии кожных проявлений (гиперкератоза, бородавках, множественных родинках) имеет смысл производить поиск мутации Ala391Glu в гене FGFR3. Для обеих форм синдрома Крузона возможна пренатальная генетическая диагностика, ультразвуковые методики при этом, как правило, малоэффективны.
|
|
Лечение
Какого-либо специфического лечения синдрома Крузона на сегодняшний момент не существует, применяют только паллиативные мероприятия. К ним относят хирургические вмешательства по ремоделированию формы черепа и устранению синостозов - такие процедуры необходимо начинать как можно раньше и в дальнейшем производить еще несколько раз по мере роста головы. Это снижает уровень внутричерепного давления, что положительно сказывается на умственном развитии больных синдромом Крузона и уменьшает вероятность появления неврологических нарушений. Также с помощью хирургических методик создают искусственный блефарофимоз для снижения степени экзофтальма и предотвращения вывиха глазного яблока. При атрезии хоан производится их расширение оперативным путем для облегчения дыхания. Описаны техники радикальных комплексных операций, направленных на устранение большинства лицевых нарушений при синдроме Крузона. В случае развития кожных изменений для снижения их выраженности рекомендуется наружное применение средств на основе ретиноидов, иногда назначают кортикостероиды.
Прогноз синдрома Крузона, как правило, неопределенный, многие специалисты оценивают его как неблагоприятный. Это связано с тем, что даже при проведении всех симптоматических и паллиативных мероприятий у больных все равно нарастает расходящееся косоглазие, практически всегда со временем развивается глухота, гипоплазия средней трети лица становится более выраженной с возрастом. Тем не менее, многие больные при соответствующем лечении и уходе могут доживать до преклонного возраста. По причине сильного нарушения зрения и слуха практически всегда происходит инвалидизация пациентов, причиной инвалидности также может стать умственная отсталость. Профилактика синдрома Крузона не разработана, возможно лишь пренатальное определение патологии молекулярно-генетическими методами.
Прогноз синдрома Крузона, как правило, неопределенный, многие специалисты оценивают его как неблагоприятный. Это связано с тем, что даже при проведении всех симптоматических и паллиативных мероприятий у больных все равно нарастает расходящееся косоглазие, практически всегда со временем развивается глухота, гипоплазия средней трети лица становится более выраженной с возрастом. Тем не менее, многие больные при соответствующем лечении и уходе могут доживать до преклонного возраста. По причине сильного нарушения зрения и слуха практически всегда происходит инвалидизация пациентов, причиной инвалидности также может стать умственная отсталость. Профилактика синдрома Крузона не разработана, возможно лишь пренатальное определение патологии молекулярно-генетическими методами.