ICD-10 codes
Description
Синдром удлиненного интервала QT. Генетически гетерогенное наследственное состояние, характеризующееся нарушением структуры и функциональности некоторых ионных каналов кардиомиоцитов. Выраженность проявлений патологии колеблется в очень широких пределах - от практически бессимптомного течения (выявляются только электрокардиологические признаки) до тяжелой глухоты, обмороков и аритмий. Определение синдрома удлиненного интервала QT производится на основании данных электрокардиологических исследований и молекулярно-генетических анализов. Лечение зависит от формы патологии и может включать в себя постоянный или курсовой прием бета-андреноблокаторов, препаратов магния и калия, а также установку дефибриллятора-кардиовертера.
Additional facts
Синдром удлиненного интервала QT - группа кардиологических расстройств генетической природы, при которых нарушается прохождение ионных токов в кардиомиоцитах, что способно приводить к аритмиям, обморокам и внезапной сердечной смерти. Впервые подобное состояние было выявлено в 1957 году норвежскими врачами А. Джервеллом и Ф. Ланге-Нильсеном, которые описали сочетание у больного врожденной глухоты, синкопальных приступов и удлинения интервала QT. Несколько позже, в 1962-64 годах были выявлены схожие симптомы у пациентов, имеющих нормальный слух - такие случаи были описаны независимо друг от друга Романо и О. Уорд. Это, а также дальнейшие открытия определили разделение синдрома удлиненного интервала QT на два клинических варианта - Романо-Уорда и Джервелла-Ланге-Нильсена. Первый наследуется по аутосомно-доминантному механизму, его частота в популяции составляет 1 случай на 5 000 населения. Встречаемость синдрома удлиненного интервала QT типа Джервелла-Ланге-Нильсена колеблется в пределах 1-6:1 000 000, он характеризуется аутосомно-доминантным путем наследования и более выраженными проявлениями. По некоторым данным, все формы синдрома удлиненного интервала QT ответственны за треть случаев внезапной сердечной смерти и около 20% внезапной младенческой смерти.
Symptoms
Ассоциированные симптомы: Судороги. Холодный пот.
Reasons
В настоящее время удалось идентифицировать 12 генов, мутации в которых приводят к развитию синдрома удлиненного интервала QT, все они кодируют те или иные белки, входящие в состав ионных каналов кардиомиоцитов, отвечающих за натриевый или калиевый ионный ток. Удалось также найти причины различий в клиническом течении этого заболевания. Аутосомно-доминантный синдром Романо-Уорда обусловлен мутацией только одного гена и поэтому может протекать бессимптомно или, как минимум, с отсутствием нарушений слуха. При типе Джервелла-Ланге-Нильсена имеется дефект двух генов - этот вариант, помимо кардиологических симптомов, всегда сопровождается двухсторонней нейросенсорной глухотой. На сегодняшний день известно, мутации каких генов обуславливают развитие синдрома удлиненного интервала QT:
1. Синдром удлиненного интервала QT тип 1 (LQT1) обусловлен мутацией гена KCNQ1, расположенного на 11-й хромосоме. Дефекты этого гена наиболее часто выявляются при наличии данного заболевания. Он кодирует последовательность альфа-субъединицы одной из разновидностей калиевых каналов кардиомиоцитов (lKs).
2. Синдром удлиненного интервала QT тип 2 (LQT2) вызывается дефектами в гене KCNH2, который локализован на 7-й хромосоме и кодирует аминокислотную последовательность белка - альфа-субъединицы другого типа калиевых каналов (lKr).
3. Синдром удлиненного интервала QT тип 3 (LQT3) обусловлен мутацией гена SCN5A, расположенного на 3-й хромосоме. В отличие от предыдущих вариантов патологии, при этом нарушается работа натриевых каналов кардиомиоцитов, так как данный ген кодирует последовательность альфа-субъединицы натриевого канала (lNa).
4. Синдром удлиненного интервала QT тип 4 (LQT4). Достаточно редкий вариант состояния, вызванный мутацией гена ANK2, который расположен на 4 - й хромосоме. Продуктом его экспрессии является белок анкирин В, который в организме человека участвует в стабилизации структуры микротрубочек миоцитов, а также выделяется в клетках нейроглии и сетчатки глаза.
5. Синдром удлиненного интервала QT тип 5 (LQT5). Разновидность заболевания, которая обусловлена дефектом в гене KCNE1, локализованном на 21 - й хромосоме. Он кодирует один из белков ионных каналов - бета-субъединицу калиевых каналов типа lKs.
6. Синдром удлиненного интервала QT типа 6 (LQT6) вызывается мутацией в гене KCNE2, расположенного также на 21-й хромосоме. Продуктом его экспрессии является бета-субъединица калиевых каналов типа lKr.
7. Синдром удлиненного интервала QT типа 7 (LQT7, другое название - синдром Андерсена, в честь педиатра Е. Д. Андерсена, описавшего это заболевание в 70-х годах) обусловлен дефектом гена KCNJ2, который локализуется на 17-й хромосоме. Как и в случае предыдущих вариантов патологии, этот ген кодирует одну из белковых цепей калиевых каналов.
8. Синдром удлиненного интервала QT типа 8 (LQT8, другое название - синдром Тимоти, в честь К. Тимоти, описавшей это заболевание) вызван мутацией гена сACNA1C, который располагается на 12-й хромосоме. Этот ген кодирует альфа-1-субъединицу кальциевого канала L-типа.
9. Синдром удлиненного интервала QT тип 9 (LQT9) обусловлен дефектом гена сAV3, локализованного на 3-й хромосоме. Продуктом его экспрессии является белок кавеолин 3, участвующий в формировании множества структур на поверхности кардиомиоцитов.
10. Синдром удлиненного интервала QT тип 10 (LQT10). Причина этой разновидности заболевания кроется в мутации гена SCN4B, который располагается на 11 - й хромосоме и отвечает за аминокислотную последовательность бета - субъединицы натриевых каналов.
11. Синдром удлиненного интервала QT тип 11 (LQT11) вызывается дефектами в гене AKAP9, расположенном на 7-й хромосоме. Он кодирует специфический белок - А-киназу центросом и комплекса Гольджи. Функции этого протеина на сегодняшний день изучены недостаточно.
12. Синдром удлиненного интервала QT тип 12 (LQT12) обусловлен мутацией гена SNTA1, локализованного на 20-й хромосоме. Он кодирует альфа-1-субъединицу белка синтрофина, участвующего в регуляции деятельности натриевых каналов кардиомиоцитов.
1. Синдром удлиненного интервала QT тип 1 (LQT1) обусловлен мутацией гена KCNQ1, расположенного на 11-й хромосоме. Дефекты этого гена наиболее часто выявляются при наличии данного заболевания. Он кодирует последовательность альфа-субъединицы одной из разновидностей калиевых каналов кардиомиоцитов (lKs).
2. Синдром удлиненного интервала QT тип 2 (LQT2) вызывается дефектами в гене KCNH2, который локализован на 7-й хромосоме и кодирует аминокислотную последовательность белка - альфа-субъединицы другого типа калиевых каналов (lKr).
3. Синдром удлиненного интервала QT тип 3 (LQT3) обусловлен мутацией гена SCN5A, расположенного на 3-й хромосоме. В отличие от предыдущих вариантов патологии, при этом нарушается работа натриевых каналов кардиомиоцитов, так как данный ген кодирует последовательность альфа-субъединицы натриевого канала (lNa).
4. Синдром удлиненного интервала QT тип 4 (LQT4). Достаточно редкий вариант состояния, вызванный мутацией гена ANK2, который расположен на 4 - й хромосоме. Продуктом его экспрессии является белок анкирин В, который в организме человека участвует в стабилизации структуры микротрубочек миоцитов, а также выделяется в клетках нейроглии и сетчатки глаза.
5. Синдром удлиненного интервала QT тип 5 (LQT5). Разновидность заболевания, которая обусловлена дефектом в гене KCNE1, локализованном на 21 - й хромосоме. Он кодирует один из белков ионных каналов - бета-субъединицу калиевых каналов типа lKs.
6. Синдром удлиненного интервала QT типа 6 (LQT6) вызывается мутацией в гене KCNE2, расположенного также на 21-й хромосоме. Продуктом его экспрессии является бета-субъединица калиевых каналов типа lKr.
7. Синдром удлиненного интервала QT типа 7 (LQT7, другое название - синдром Андерсена, в честь педиатра Е. Д. Андерсена, описавшего это заболевание в 70-х годах) обусловлен дефектом гена KCNJ2, который локализуется на 17-й хромосоме. Как и в случае предыдущих вариантов патологии, этот ген кодирует одну из белковых цепей калиевых каналов.
8. Синдром удлиненного интервала QT типа 8 (LQT8, другое название - синдром Тимоти, в честь К. Тимоти, описавшей это заболевание) вызван мутацией гена сACNA1C, который располагается на 12-й хромосоме. Этот ген кодирует альфа-1-субъединицу кальциевого канала L-типа.
9. Синдром удлиненного интервала QT тип 9 (LQT9) обусловлен дефектом гена сAV3, локализованного на 3-й хромосоме. Продуктом его экспрессии является белок кавеолин 3, участвующий в формировании множества структур на поверхности кардиомиоцитов.
10. Синдром удлиненного интервала QT тип 10 (LQT10). Причина этой разновидности заболевания кроется в мутации гена SCN4B, который располагается на 11 - й хромосоме и отвечает за аминокислотную последовательность бета - субъединицы натриевых каналов.
11. Синдром удлиненного интервала QT тип 11 (LQT11) вызывается дефектами в гене AKAP9, расположенном на 7-й хромосоме. Он кодирует специфический белок - А-киназу центросом и комплекса Гольджи. Функции этого протеина на сегодняшний день изучены недостаточно.
12. Синдром удлиненного интервала QT тип 12 (LQT12) обусловлен мутацией гена SNTA1, локализованного на 20-й хромосоме. Он кодирует альфа-1-субъединицу белка синтрофина, участвующего в регуляции деятельности натриевых каналов кардиомиоцитов.
Classification
Несмотря на широкое генетическое разнообразие синдрома удлиненного интервала QT, общие звенья его патогенеза в целом одинаковы для каждой из форм. Данное заболевание относят к группе каналопатий из-за того, что его причиной выступают нарушения в строении тех или иных ионных каналов. В результате этого процессы реполяризации миокарда происходят неравномерно и не одновременно в различных частях желудочков, что становится причиной удлинения интервала QT. Кроме того, значительно возрастает чувствительность миокарда к влияниям симпатической нервной системы, что становится причиной частых тахиаритмий, способных приводить к жизнеугрожающим фибрилляциям желудочков. При этом у разных генетических типов синдрома удлиненного интервала QT отмечается различная чувствительность к тем или иным воздействиям. Например, LQT1 характеризуется синкопальными приступами и аритмией при физической нагрузке, при LQT2 аналогичные проявления наблюдаются при громких и резких звуках, для LQT3, напротив, более характерно развитие аритмий и фибрилляций в спокойном состоянии (например, во сне).
Diagnostics
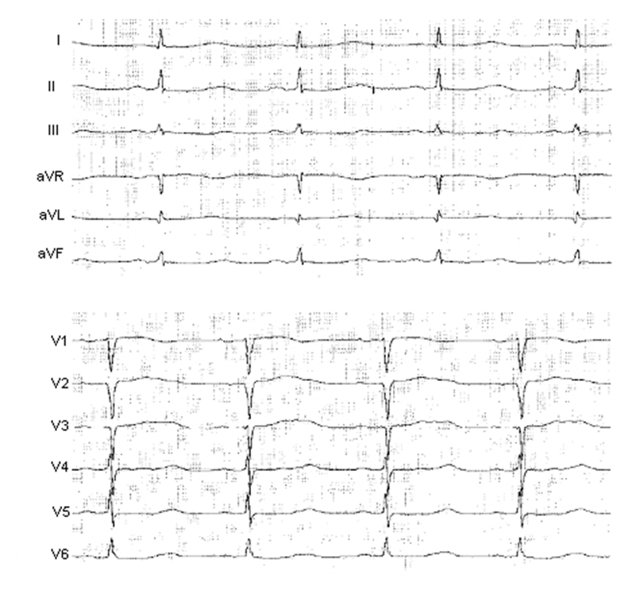
Диагностика синдрома удлиненного интервала QT производится на основании изучения анамнеза больного, электрокардиологических и молекулярно-генетических исследований. При расспросе пациента часто обнаруживаются эпизоды обмороков, головокружений, ощущения сердцебиений, но при легких формах патологии их может и не быть. Иногда аналогичные проявления встречаются у кого-либо из родственников пациента, что указывает на семейный характер заболевания. При любой форме синдрома удлиненного интервала QT будут выявляться изменения на ЭКГ - увеличение интервала QT до 0,6 секунд и более, возможно увеличение амплитуды зубца Т. Сочетание таких ЭКГ-признаков с врожденной глухотой говорит о наличии синдрома Джервелла-Ланге-Нильсена. Кроме того, часто необходимо холтеровское мониторирование работы сердца на протяжении суток для выявления возможных приступов тахиаритмий. Определение синдрома удлиненного интервала QT при помощи методов современной генетики на сегодняшний день возможно в отношении практических всех генетических типов этого заболевания.
Treatment
Терапия синдрома удлиненного интервала QT достаточно сложна, многие специалисты рекомендуют при этом заболевании одни схемы и отвергают другие, но какого-либо единого протокола лечения этой патологии не существует. Универсальными препаратами считаются бета-адреноблокаторы, которые уменьшают риск развития тахиаритмий и фибрилляций, а также снижают степень симпатических воздействий на миокард, но при LQT3 они малоэффективны. В случае синдрома удлиненного интервала QT типа 3 более разумно использовать антиаритмические препараты класса В1. Эти особенности лечения заболевания повышают потребность в молекулярно-генетической диагностике для определения типа патологии. В случае частых приступов тахиаритмий и высокого риска развития фибрилляции рекомендуется имплантация кардиостимулятора или дефибриллятора-кардиовертера.