
Description
Синдром Кальмана. Это наследственное заболевание, проявляющееся аносмией и гипогонадотропным гипогонадизмом. Патология вызывается несколькими типами мутаций в Х-хромосоме или аутосомах. Синдром проявляется недоразвитием первичных и вторичных половых признаков, нарушением способности различать запахи, задержкой психомоторного развития. Для постановки диагноза проводят гормональные и генетические исследования, УЗИ органов малого таза у женщин и мошонки у мужчин, магнитно-резонансную томографию. Лечение болезни Кальмана включает пожизненную заместительную гормональную терапию, хирургическую коррекцию сопутствующих пороков.
Additional facts
Сочетание задержки полового развития и нарушения обоняния подробно описал американский психолог и генетик Ф.Дж. Кальман в 1944 г. в научной работе «Генетические аспекты первичного евнухоидизма». Однако первое упоминание о болезни относится к 1856 году и связано с именем испанского врача Аурелиано Маэстре де Сан-Хуана. Впоследствии патологию назвали синдромом Калмана (Kallman s) или ольфактогенитальной дисплазией. Встречается с частотой 1:10 000 у новорожденных и 1:50 000 у девочек.
Reasons
Патология имеет наследственное происхождение, возникает под влиянием точечных генетических мутаций. Заболевание встречается в виде спорадических случаев или в семейной форме. Передача мутантного гена происходит по Х-сцепленному аутосомно-доминантному рецессивному типу наследования. В зависимости от характера изменений генетического материала в генетике выделяют 3 типичные формы заболевания:
• Мутация KALIG 1. Патологический ген расположен на Х-хромосоме и вызывает синдром Кальмана типа 1. Помимо основных признаков, наблюдаются множественные аномалии развития почек.
• Мутация KAL 2. Изменения происходят на коротком плече 8-й хромосомы, что приводит к 2-му типу обонятельной дисплазии. Патогномоничные признаки дополняются нарушением слуха, низкорослостью и умственной отсталостью.
• Мутация KAL 3. Возникает при патологии генов 20-й хромосомы, является причиной 3-го типа болезни Кальмана. Характеризуется множественными пороками развития, в том числе поражением лицевого скелета.
• Мутация KALIG 1. Патологический ген расположен на Х-хромосоме и вызывает синдром Кальмана типа 1. Помимо основных признаков, наблюдаются множественные аномалии развития почек.
• Мутация KAL 2. Изменения происходят на коротком плече 8-й хромосомы, что приводит к 2-му типу обонятельной дисплазии. Патогномоничные признаки дополняются нарушением слуха, низкорослостью и умственной отсталостью.
• Мутация KAL 3. Возникает при патологии генов 20-й хромосомы, является причиной 3-го типа болезни Кальмана. Характеризуется множественными пороками развития, в том числе поражением лицевого скелета.
Pathogenesis
В основе заболевания лежит генетически детерминированный дефицит гонадотропин-рилизинг-гормонов в гипоталамусе. Мутантные гены участвуют в миграции нейронов, ответственных за синтез и выделение гормональных веществ, поэтому при ольфактогенитальной дисплазии этот процесс нарушается. В результате уменьшается выброс лютеинизирующего и фолликулостимулирующего гормонов из гипофиза, развивается вторичный врожденный гипогонадизм.
Аносмия при синдроме Кальмана связана с внутриутробной гипоплазией или аплазией обонятельных луковиц. Считается, что мутантные аллели нарушают нормальное расположение нейронов в обонятельных структурах, что изменяет способность воспринимать запахи. Пока неизвестно, как генетические аномалии влияют на возникновение других врожденных дефектов, поэтому подозреваются дополнительные наследственные факторы и факторы окружающей среды.
Синдром Кальмана.
Аносмия при синдроме Кальмана связана с внутриутробной гипоплазией или аплазией обонятельных луковиц. Считается, что мутантные аллели нарушают нормальное расположение нейронов в обонятельных структурах, что изменяет способность воспринимать запахи. Пока неизвестно, как генетические аномалии влияют на возникновение других врожденных дефектов, поэтому подозреваются дополнительные наследственные факторы и факторы окружающей среды.
Синдром Кальмана.

Symptoms
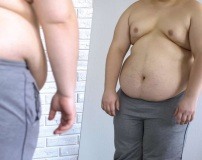
Клинические проявления заболевания вариабельны, даже у членов одной семьи могут быть разные по степени тяжести формы обонятельно-генитальной дисплазии. Типичные проявления заболевания в основном отмечаются в пубертатном периоде, когда становится заметной задержка полового развития. У мальчиков формируется евнухоидизм, проявляющийся недоразвитием мошонки и яичек, малыми размерами полового члена и отсутствием оволосения в гормонозависимых зонах.
Для женщин характерно недоразвитие половых признаков: отсутствие округлости молочных желез в период полового созревания, скудное оволосение на теле, непропорциональность фигуры и нарушения отложения жировой ткани. У подростков первичная аменорея. Внутренние половые органы у женщин также гипопластичны, размеры матки и яичников малы, нарушена секреция эстрогенов и прогестерона.
Обонятельные патологии варьируют от незначительного снижения способности распознавать запахи до полной аносмии. До 13-14 лет больные синдромом Кальмана не отстают от своих сверстников по параметрам тела, однако у них не наблюдается пубертатного скачка роста и снижаются темпы роста. Часто наблюдается задержка дифференцировки скелетных структур, из-за чего костный возраст больного будет ниже паспортного.
Ассоциированные симптомы: Нарушение обоняния. Потеря обоняния.
Для женщин характерно недоразвитие половых признаков: отсутствие округлости молочных желез в период полового созревания, скудное оволосение на теле, непропорциональность фигуры и нарушения отложения жировой ткани. У подростков первичная аменорея. Внутренние половые органы у женщин также гипопластичны, размеры матки и яичников малы, нарушена секреция эстрогенов и прогестерона.
Обонятельные патологии варьируют от незначительного снижения способности распознавать запахи до полной аносмии. До 13-14 лет больные синдромом Кальмана не отстают от своих сверстников по параметрам тела, однако у них не наблюдается пубертатного скачка роста и снижаются темпы роста. Часто наблюдается задержка дифференцировки скелетных структур, из-за чего костный возраст больного будет ниже паспортного.
Ассоциированные симптомы: Нарушение обоняния. Потеря обоняния.
Possible complications
Примерно у 5-10% больных с синдромом Кальмана имеются сопутствующие пороки развития внутренних органов и скелета. Типичными осложнениями заболевания являются расщелина губы, готическое небо, агенезия почек. К неврологическим последствиям обонятельной дисплазии относятся двусторонние синкинезии, атаксия, умственная отсталость различной степени тяжести. В редких случаях клиническая картина дополняется пороками сердца.
Серьезной проблемой для больных является нарушение полового созревания. Мужской гипогонадизм сопровождается микропенисом, снижением продукции тестостерона и снижением секреции спермы, что вызывает объективные и психологические трудности при попытке полового акта. У женщин развивается первичное бесплодие, стойкое снижение либидо.
Серьезной проблемой для больных является нарушение полового созревания. Мужской гипогонадизм сопровождается микропенисом, снижением продукции тестостерона и снижением секреции спермы, что вызывает объективные и психологические трудности при попытке полового акта. У женщин развивается первичное бесплодие, стойкое снижение либидо.

Diagnostics
Обследование больных с подозрением на синдром Кальмана проводит врач-эндокринолог с участием невролога, генетика и других специалистов, при необходимости. При физикальном обследовании определяют формулу полового развития, соответствие физических параметров тела возрасту больного, наличие видимых признаков врожденных пороков развития. Диагностика обонятельной дисплазии комплексная и включает следующие методы исследования:
• УЗИ половых органов. Женщинам проводят УЗИ органов малого таза для изучения размеров матки и придатков для оценки степени гипоплазии яичников. У мужчин проводят УЗИ мошонки, которое определяет значительное уменьшение объема яичек при их однородной структуре и правильной эхогенности.
• Ольфактометрия. Для оценки степени уменьшения запаха проводится стандартный тест набора ароматизаторов. Дополнительно проводят риноскопию и рентгенографию придаточных пазух носа для исключения риногенной причины аносмии.
• МРТ головного мозга. Выраженных структурных изменений головного мозга при синдроме Кальмана нет. Изредка определяют неоднородность строения аденогипофиза, признаки гипоксической энцефалопатии и другие неспецифические неврологические признаки.
• Гормональный профиль. Определение уровня половых гормонов имеет диагностическое значение в пубертатном периоде, когда в норме происходит половое созревание. У мальчиков определяют снижение уровня тестостерона, у девочек выявляют недостаток эстрогенов. Независимо от пола развивается стойкое снижение уровня рилизинг-гормонов гипоталамуса и гипофизарных гонадотропинов.
• Генетический анализ. Определение характерных мутаций - единственный 100% метод подтверждения диагноза синдрома Кальмана. Все пациенты с типичными клиническими проявлениями направляются на консультацию генетика с целью дифференциации ольфактогенитального синдрома от других форм врожденной патологии.
• УЗИ половых органов. Женщинам проводят УЗИ органов малого таза для изучения размеров матки и придатков для оценки степени гипоплазии яичников. У мужчин проводят УЗИ мошонки, которое определяет значительное уменьшение объема яичек при их однородной структуре и правильной эхогенности.
• Ольфактометрия. Для оценки степени уменьшения запаха проводится стандартный тест набора ароматизаторов. Дополнительно проводят риноскопию и рентгенографию придаточных пазух носа для исключения риногенной причины аносмии.
• МРТ головного мозга. Выраженных структурных изменений головного мозга при синдроме Кальмана нет. Изредка определяют неоднородность строения аденогипофиза, признаки гипоксической энцефалопатии и другие неспецифические неврологические признаки.
• Гормональный профиль. Определение уровня половых гормонов имеет диагностическое значение в пубертатном периоде, когда в норме происходит половое созревание. У мальчиков определяют снижение уровня тестостерона, у девочек выявляют недостаток эстрогенов. Независимо от пола развивается стойкое снижение уровня рилизинг-гормонов гипоталамуса и гипофизарных гонадотропинов.
• Генетический анализ. Определение характерных мутаций - единственный 100% метод подтверждения диагноза синдрома Кальмана. Все пациенты с типичными клиническими проявлениями направляются на консультацию генетика с целью дифференциации ольфактогенитального синдрома от других форм врожденной патологии.
Dif. diagnostics
При выявлении гипогонадизма проводят дифференциацию его первичной, вторичной и третичной форм, что требует проведения функциональных проб с ГнРГ и кломифеном. В группе больных до 18 лет необходимо исключить конституциональную задержку полового созревания, которая может имитировать легкий вариант синдрома Кальмана. Дифференциальную диагностику аносмии проводят с опухолевыми и другими органическими поражениями ЦНС.
Диагностика синдрома Кальмана.
Диагностика синдрома Кальмана.
Treatment
Основная цель лечения - стимуляция полового развития с помощью индивидуальных схем гормонотерапии. Терапию начинают в пубертатном периоде с 13-13,5 лет и проводят пожизненно. Дополнительно назначают препараты холекальциферола и витаминные комплексы, препятствующие развитию остеопороза. Аносмия у больных не подлежит коррекции из-за отсутствия эффективных методов терапии.
Для лечения бесплодия у женщин применяют хорионический гонадотропин в сочетании с гонадолиберином или менотропином, которые назначают в импульсном режиме в течение нескольких лет. В период подготовки к беременности используется гормональная стимуляция овуляции, при этом вероятность физиологического зачатия составляет более 70%. У мужчин проводят заместительную терапию гонадолиберинами и тестостероном.
У взрослых мужчин с синдромом Кальмана и микропенисом возможна хирургическая коррекция - лигаментотомия, которая направлена на удлинение полового члена и улучшение половой функции. При наличии врожденных аномалий развития лица в соответствующие сроки проводится их пластическая коррекция. Лечение пороков сердца проводят кардиохирурги в соответствии с действующими протоколами.
Для лечения бесплодия у женщин применяют хорионический гонадотропин в сочетании с гонадолиберином или менотропином, которые назначают в импульсном режиме в течение нескольких лет. В период подготовки к беременности используется гормональная стимуляция овуляции, при этом вероятность физиологического зачатия составляет более 70%. У мужчин проводят заместительную терапию гонадолиберинами и тестостероном.
У взрослых мужчин с синдромом Кальмана и микропенисом возможна хирургическая коррекция - лигаментотомия, которая направлена на удлинение полового члена и улучшение половой функции. При наличии врожденных аномалий развития лица в соответствующие сроки проводится их пластическая коррекция. Лечение пороков сердца проводят кардиохирурги в соответствии с действующими протоколами.
Forecast
Качество жизни больных зависит от формы синдрома Кальмана и тяжести его проявлений. Легкие и субклинические варианты заболевания имеют благоприятный прогноз, при гормональной коррекции удается добиться нормального полового развития и репродуктивной функции. Менее оптимистичен прогноз при выраженной обонятельной дисплазии в сочетании с другими врожденными пороками развития. В связи со сложностью этиопатогенеза меры профилактики до сих пор не разработаны.
References
1. Синдром Кальмана у мальчика 17 лет/ А.В. Бурлуцкая, О.Г. Коробкина, А.В. Статова// Кубанский научный медицинский вестник. 2020. №1.
2. Аспекты диагностики и терапии синдрома Кальмана у мальчика (случай из практики) / О.А. Оганова/ Медицинский совет. 2013. №1.
3. Kallmann syndrome / Dodé, сatherine, and Jean-Pierre Hardelin // European Journal of Human Genetics. 2009. №2.
4. Случай ольфактогенитальной дисплазии (синдрома Каллмана) у женщины / М.Б. Бабарина, А.В. Секинаева, Е.Н. Гиниятуллина, Л.Я. Рожинская // Проблемы эндокринологии. 2006. №1.
2. Аспекты диагностики и терапии синдрома Кальмана у мальчика (случай из практики) / О.А. Оганова/ Медицинский совет. 2013. №1.
3. Kallmann syndrome / Dodé, сatherine, and Jean-Pierre Hardelin // European Journal of Human Genetics. 2009. №2.
4. Случай ольфактогенитальной дисплазии (синдрома Каллмана) у женщины / М.Б. Бабарина, А.В. Секинаева, Е.Н. Гиниятуллина, Л.Я. Рожинская // Проблемы эндокринологии. 2006. №1.