ICD-10 codes
Description
Потомственный Джейд. Это генетически обусловленная неиммунная гломерулопатия, обычно приводящая к почечной недостаточности. Это проявляется астеническими синдромами, интоксикацией, задержкой физического развития у детей, макрогематурией, полиурией, никтурией, отеками, артериальной гипертонией. Поставлен диагноз: общий анализ мочи, пункционная биопсия, электронная микроскопия. Рекомендуется симптоматическая терапия ингибиторами АПФ, блокаторами рецепторов ангиотензина, иммуномодуляторами, анаболиками и ингибиторами кальциневрина. При терминальной почечной недостаточности показаны заместительная терапия и трансплантация почки.
Additional facts
Распространенность наследственного нефрита, по данным зарубежных экспертов в области нефрологии, составляет 0,01-0,02%. В России заболевание выявляется у 0,017% детей. Первые наблюдения за пациентами с семейными формами гломерулопатии, приводящими к уремии, были сделаны в 1902-1923 гг.
В 1927 году британский ученый Артур Альпорт выявил вариант наследственной комбинации уремии и потери слуха, который впоследствии был назван в его честь - синдром Альпорта. Генетическая основа наследственных вариантов нефрита была установлена в 1985 году. В более чем 80% случаев заболевание начинается в возрасте от 3 до 10 лет и является более тяжелым у пациентов мужского пола из-за преобладания л наследование связано с X.
В 1927 году британский ученый Артур Альпорт выявил вариант наследственной комбинации уремии и потери слуха, который впоследствии был назван в его честь - синдром Альпорта. Генетическая основа наследственных вариантов нефрита была установлена в 1985 году. В более чем 80% случаев заболевание начинается в возрасте от 3 до 10 лет и является более тяжелым у пациентов мужского пола из-за преобладания л наследование связано с X.
Reasons
Заболевание является наследственным и связано с генетическим дефектом, при котором нарушается биосинтез коллагеновых волокон типа IV, которые являются частью базальных мембран нефронов и многих других органов. Несколько генов (C0L4A5, с0L4A3, с0L4A4), расположенных на разных хромосомах, ответственны за кодирование цепей коллагена, которые образуют клубочковые мембраны.
Наследственные формы нефрита являются генетически неоднородными, что влияет на выраженность клинических симптомов, скорость развития заболевания, время, когда происходит декомпенсация. Чаще всего ген сOL4A5, кодирующий α-5 цепь коллагеновых волокон, удаляется, точечные мутации и нарушения сплайсинга. У 80% пациентов заболевание наследуется по Х-сцепленному типу, у 15% - по аутосомно-рецессивному, у 5% - по аутосомно-доминантному.
Наследственные формы нефрита являются генетически неоднородными, что влияет на выраженность клинических симптомов, скорость развития заболевания, время, когда происходит декомпенсация. Чаще всего ген сOL4A5, кодирующий α-5 цепь коллагеновых волокон, удаляется, точечные мутации и нарушения сплайсинга. У 80% пациентов заболевание наследуется по Х-сцепленному типу, у 15% - по аутосомно-рецессивному, у 5% - по аутосомно-доминантному.
Pathogenesis
Механизм развития патологических изменений при наследственном нефрите основан на нарушении нормальной структуры базальных мембран в тканях почек и некоторых других органов. Мутация в гене с0L4A5, который расположен в локусе Xq21.3 на длинном плече Х-хромосомы, изменяет структуру цепи α-5 коллагена типа 4. Повреждение генов с0L4A3, с0L4A4, расположенных на 2-й хромосоме сопровождаются нарушением синтеза α-3 и α-4 цепей коллагеновых волокон.
Аномалии одной из этих цепей влияют на формирование базальных мембран клубочков, дистальных канальцев и собирательных протоков. Из-за более плотного многослойного плетения или неправильного пространственного распределения волокон мембраны в основании утолщаются, отслаиваются и истончаются. В почечных органах происходит пролиферация мезангиоцитов, накапливается мезангиальный матрикс, клубочки склерозируются, а местный гломерулосклероз сменяется сегментарным гломерулосклерозом, глобальным и усугубляемым гиалинозом.
В то же время развивается атрофия канальцев, развивается интерстициальный фиброз, помимо нормальных интерстициальных клеток появляются пенистые клеточные элементы. Патоморфологические изменения клинически проявляются нарушением функций фильтрации и реабсорбции. Поскольку сходные коллагеновые волокна являются частью специфических мембран хрусталика и кортиева органа, у пациентов, помимо урологической патологии, отмечаются наследственные формы нарушения зрения и слуха.
Аномалии одной из этих цепей влияют на формирование базальных мембран клубочков, дистальных канальцев и собирательных протоков. Из-за более плотного многослойного плетения или неправильного пространственного распределения волокон мембраны в основании утолщаются, отслаиваются и истончаются. В почечных органах происходит пролиферация мезангиоцитов, накапливается мезангиальный матрикс, клубочки склерозируются, а местный гломерулосклероз сменяется сегментарным гломерулосклерозом, глобальным и усугубляемым гиалинозом.
В то же время развивается атрофия канальцев, развивается интерстициальный фиброз, помимо нормальных интерстициальных клеток появляются пенистые клеточные элементы. Патоморфологические изменения клинически проявляются нарушением функций фильтрации и реабсорбции. Поскольку сходные коллагеновые волокна являются частью специфических мембран хрусталика и кортиева органа, у пациентов, помимо урологической патологии, отмечаются наследственные формы нарушения зрения и слуха.
Classification
При систематизации форм наследственного нефрита учитываются характер и степень выраженности клинических проявлений нефрологической патологии, динамика развития заболевания, время начала почечной недостаточности, наличие признаков повреждения других органов. Наличие нескольких вариантов патологии определяется типом и степенью экспрессии мутированных генов. Специалисты в области клинической урологии и медицинской генетики выделяют следующие формы наследственной нефропатии:
Ювенильный нефрит с поражением почек, потерей слуха и нарушением зрения (синдром Альпорта). Это проявляется, когда ген с0L4A5 поврежден (доминантное наследование связано с Х-хромосомой). Проявляется рано, отличается прогрессирующим течением с развитием хронической почечной недостаточности. У 50% пациентов терминальная стадия почечной недостаточности наступает до 25 лет, у 90% - до 40 лет.
Наследственная форма нефрита без потери слуха. Он передается по аутосомно-рецессивному механизму. Мутации обнаруживаются в двух аллелях генов сOL4A3 или сOL4A4. В клинической картине появляются почечные симптомы. Заболевание имеет прогрессирующее течение, терминальная стадия заболевания почек формируется к 30 годам.
Доброкачественная семейная гематурия. Он унаследован от аутосомно-доминантного типа и связан с мутацией в одном из генов 2-й хромосомы (COL4A3 или сOL4A4). Появляется позже, иногда в зрелом возрасте. Он характеризуется медленным прогрессированием с умеренными нефрологическими симптомами и низкой вероятностью развития хронической почечной недостаточности.
Ювенильный нефрит с поражением почек, потерей слуха и нарушением зрения (синдром Альпорта). Это проявляется, когда ген с0L4A5 поврежден (доминантное наследование связано с Х-хромосомой). Проявляется рано, отличается прогрессирующим течением с развитием хронической почечной недостаточности. У 50% пациентов терминальная стадия почечной недостаточности наступает до 25 лет, у 90% - до 40 лет.
Наследственная форма нефрита без потери слуха. Он передается по аутосомно-рецессивному механизму. Мутации обнаруживаются в двух аллелях генов сOL4A3 или сOL4A4. В клинической картине появляются почечные симптомы. Заболевание имеет прогрессирующее течение, терминальная стадия заболевания почек формируется к 30 годам.
Доброкачественная семейная гематурия. Он унаследован от аутосомно-доминантного типа и связан с мутацией в одном из генов 2-й хромосомы (COL4A3 или сOL4A4). Появляется позже, иногда в зрелом возрасте. Он характеризуется медленным прогрессированием с умеренными нефрологическими симптомами и низкой вероятностью развития хронической почечной недостаточности.
Symptoms
На ранних стадиях заболевания клиническая картина характеризуется общими нарушениями: замедлением роста и физического развития, синдромом интоксикации, который приводит к слабости, усталости, головокружению, бледности и сухости кожи, плохому аппетиту, снижению мышечного тонуса, частым головным болям, шуму в ушах и бессоннице манифесты.
По мере прогрессирования нефрита появляются такие симптомы, как частое мочеиспускание, увеличение мочеиспускания по ночам, боль в области живота или таза, появление утреннего отека, видимая кровь в моче (макрогематурия) и постоянное повышение артериального давления. Кроме того, у пациента с наследственной гломерулопатией может быть выявлено клеймо (небольшие нарушения в эмбриогенезе) - деформация ушных раковин, эпикантуса, высокого неба, асимметрия грудной клетки, синдактилия (слияние двух или более пальцев).
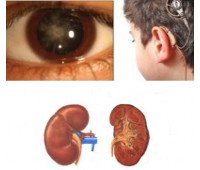
У 30-50% пациентов наблюдаются признаки нарушения слуха - от небольших изменений, обнаруженных на аудиограмме, до полной глухоты. В 15-30% случаев диагностируются аномалии в строении хрусталика и нарушения зрения в виде передней чечевицы, миопия, гиперметропия, астигматизм, катаракта, кератоконус, сферофация, пигментный ретинит, амавроз, нистагм и ;
Ассоциированные симптомы: Гематурия. Лейкоцитурия. Недомогание. Протеинурия. Цилиндрурия. Шум в ушах.
По мере прогрессирования нефрита появляются такие симптомы, как частое мочеиспускание, увеличение мочеиспускания по ночам, боль в области живота или таза, появление утреннего отека, видимая кровь в моче (макрогематурия) и постоянное повышение артериального давления. Кроме того, у пациента с наследственной гломерулопатией может быть выявлено клеймо (небольшие нарушения в эмбриогенезе) - деформация ушных раковин, эпикантуса, высокого неба, асимметрия грудной клетки, синдактилия (слияние двух или более пальцев).
У 30-50% пациентов наблюдаются признаки нарушения слуха - от небольших изменений, обнаруженных на аудиограмме, до полной глухоты. В 15-30% случаев диагностируются аномалии в строении хрусталика и нарушения зрения в виде передней чечевицы, миопия, гиперметропия, астигматизм, катаракта, кератоконус, сферофация, пигментный ретинит, амавроз, нистагм и ;
Ассоциированные симптомы: Гематурия. Лейкоцитурия. Недомогание. Протеинурия. Цилиндрурия. Шум в ушах.
Possible complications
При длительном течении наследственного нефрита происходит постепенная гибель нефронов, что обычно осложняет заболевание с увеличением почечной недостаточности. При юношеских формах нефропатии хроническая почечная недостаточность возникает в возрасте от 16 до 20 лет и, при недостаточном лечении, приводит к смерти пациента в возрасте до 30 лет. Генетический нефрит, сопровождающийся метаболическими нарушениями, может привести к чрезмерному образованию соли и отложению, что вызывает развитие мочекаменной болезни. При увеличении функциональной нагрузки на почки возможно развитие острой почечной недостаточности с необходимостью экстренной терапии.
Diagnostics
Точное генеалогическое исследование играет важную роль в диагностике наследственного нефрита. У 80% пациентов можно выявить семейные формы урологической патологии, наследственную потерю слуха и зрения с более тяжелыми симптомами у мужчин. При предполагаемой связи заболевания с генетическими отклонениями рекомендуются методы исследования, позволяющие выявить признаки повреждения базальных мембран, функциональную почечную недостаточность, морфологические изменения в паренхиме, характерные для наследственных форм нефрита:
• Общий анализ мочи. По меньшей мере, в двух образцах мочи, собранных в разное время, измененные эритроциты обнаруживаются в 3 единицах в поле зрения или более. Характерны протеинурия (более 0,35 г / л), лейкоцитурия, цилиндрурия, которые усиливаются по мере развития заболевания.
• Биопсия почки путем укола. При гистологическом исследовании почечных биопсий в канальцах выявляется накопление эритроцитов, инфильтрат интерстициальной ткани, клубочки сосредоточены. Иммунофлуоресцентный анализ выявляет атипичный коллаген типа 4.
• Электронная микроскопия. На ранних стадиях нефрита клубочковые базальные мембраны истончены; на более поздних стадиях они становятся тоньше или толще, они становятся слоистыми, разделенными. Пролиферация мезангиоцитов, характерно расширение мезангиев.
В настоящее время методы идентификации мутантного гена используются редко из-за технической сложности и высокой стоимости исследования. Изменения в биохимии крови (повышение уровня креатинина, мочевой кислоты, азота мочевины, снижение общего белка) и соответствующие изменения в анализе мочи (положительные тесты на почки) свидетельствуют о развитии почечной недостаточности, которая осложняет течение наследственного нефрита.
Заболевание дифференцируется с паранеопластической нефропатией, туберкулезом почек, острым и хроническим гломерулонефритом, пиелонефритом, мочекаменной болезнью, подагрическим интерстициальным нефритом, гломерулярными поражениями при системных заболеваниях соединительной ткани. В дополнение к осмотру нефролога или уролога пациенту рекомендуется проконсультироваться с медицинским генетиком по показаниям: отоларинголог, офтальмолог, врач общей практики, эндокринолог, ревматолог, иммунолог, онколог и гематолог.
• Общий анализ мочи. По меньшей мере, в двух образцах мочи, собранных в разное время, измененные эритроциты обнаруживаются в 3 единицах в поле зрения или более. Характерны протеинурия (более 0,35 г / л), лейкоцитурия, цилиндрурия, которые усиливаются по мере развития заболевания.
• Биопсия почки путем укола. При гистологическом исследовании почечных биопсий в канальцах выявляется накопление эритроцитов, инфильтрат интерстициальной ткани, клубочки сосредоточены. Иммунофлуоресцентный анализ выявляет атипичный коллаген типа 4.
• Электронная микроскопия. На ранних стадиях нефрита клубочковые базальные мембраны истончены; на более поздних стадиях они становятся тоньше или толще, они становятся слоистыми, разделенными. Пролиферация мезангиоцитов, характерно расширение мезангиев.
В настоящее время методы идентификации мутантного гена используются редко из-за технической сложности и высокой стоимости исследования. Изменения в биохимии крови (повышение уровня креатинина, мочевой кислоты, азота мочевины, снижение общего белка) и соответствующие изменения в анализе мочи (положительные тесты на почки) свидетельствуют о развитии почечной недостаточности, которая осложняет течение наследственного нефрита.
Заболевание дифференцируется с паранеопластической нефропатией, туберкулезом почек, острым и хроническим гломерулонефритом, пиелонефритом, мочекаменной болезнью, подагрическим интерстициальным нефритом, гломерулярными поражениями при системных заболеваниях соединительной ткани. В дополнение к осмотру нефролога или уролога пациенту рекомендуется проконсультироваться с медицинским генетиком по показаниям: отоларинголог, офтальмолог, врач общей практики, эндокринолог, ревматолог, иммунолог, онколог и гематолог.
Treatment
Эффективной этиопатогенетической терапии заболевания не предлагается. Администрация гормонов и иммунодепрессантов обычно неэффективна. Ренопротекция рекомендуется пациентам с наследственной формой нефрита: низкобелковая диета, ограничение физической активности, восстановление очагов хронической инфекции, осторожность при проведении плановых прививок и назначении препаратов с нефротоксичностью. Для предотвращения раннего развития интерстициального фиброза назначают активную антипротеинурическую терапию для предотвращения повреждения и атрофии эпителиальных клеток почечных канальцев:
• Ингибиторы ангиотензин-превращающего фермента (ингибитор АПФ). Это препараты первой линии; вызывают расширение кровеносных сосудов, улучшают почечный кровоток, снижают повышенное внутричерепное давление, уменьшают реабсорбцию натрия и воды. Благодаря ингибированию ангиотензина II они оказывают антифиброгенное действие и ингибируют развитие тубулоинтерстициального фиброза.
• Блокаторы рецепторов ангиотензина (ARBs). Используется в качестве второй строки носителя. Рецепторы ангиотензина II AT-1 связаны между собой, блокируя его действие. Хотя почечные эффекты БРА, как правило, аналогичны ингибиторам АПФ, препараты этой группы влияют более мягко и на специальной основе, не влияют на систему ангиотензинпревращающего фермента, ионные каналы и другие приемники.
Антипротеинурическую терапию дополняют витаминные комплексы, препараты, аналогичные витаминам, препараты с иммуномодулирующим и анаболическим действием, ингибиторы кальциневрина, гипербарическая оксигенация. Появление признаков терминальной стадии почечной недостаточности служит показанием для заместительной почечной терапии - гемодиализ, перитонеальный диализ, гемофильтрация.
Единственный способ радикально улучшить состояние пациента - это пересадка почки, хотя такая операция имеет определенные показания и противопоказания по сравнению с другими типами нефрологической патологии. При выборе относительного донора следует иметь в виду, что он также может иметь бессимптомную наследственную предрасположенность к нефриту, который декомпрессируется после удаления одной из почек. Кроме того, наличие нормальной альфа-цепи коллагена, присутствующего в базальных мембранах трансплантированного органа, может вызвать иммунный ответ и отторжение почки.
• Ингибиторы ангиотензин-превращающего фермента (ингибитор АПФ). Это препараты первой линии; вызывают расширение кровеносных сосудов, улучшают почечный кровоток, снижают повышенное внутричерепное давление, уменьшают реабсорбцию натрия и воды. Благодаря ингибированию ангиотензина II они оказывают антифиброгенное действие и ингибируют развитие тубулоинтерстициального фиброза.
• Блокаторы рецепторов ангиотензина (ARBs). Используется в качестве второй строки носителя. Рецепторы ангиотензина II AT-1 связаны между собой, блокируя его действие. Хотя почечные эффекты БРА, как правило, аналогичны ингибиторам АПФ, препараты этой группы влияют более мягко и на специальной основе, не влияют на систему ангиотензинпревращающего фермента, ионные каналы и другие приемники.
Антипротеинурическую терапию дополняют витаминные комплексы, препараты, аналогичные витаминам, препараты с иммуномодулирующим и анаболическим действием, ингибиторы кальциневрина, гипербарическая оксигенация. Появление признаков терминальной стадии почечной недостаточности служит показанием для заместительной почечной терапии - гемодиализ, перитонеальный диализ, гемофильтрация.
Единственный способ радикально улучшить состояние пациента - это пересадка почки, хотя такая операция имеет определенные показания и противопоказания по сравнению с другими типами нефрологической патологии. При выборе относительного донора следует иметь в виду, что он также может иметь бессимптомную наследственную предрасположенность к нефриту, который декомпрессируется после удаления одной из почек. Кроме того, наличие нормальной альфа-цепи коллагена, присутствующего в базальных мембранах трансплантированного органа, может вызвать иммунный ответ и отторжение почки.
References
1. Детская нефрология. Практическое руководство/ Под редакцией Лойманна Э., Цыгина А.Н., Саркисяна А.А. 2010.
2. Характеристика наследственного нефрита у детей/ Белькевич А.Г., Козыро И.А., Сукало А.В. Актуальные вопросы педиатрии: сборник материалов межрегиональной научно-практической конференции с международным участием, Гродно, 19-20 апреля 2018 г.
3. Наследственные заболевания почек, протекающие с гематурией/ Игнатова М.С., Длин В.В. Российский вестник перинатологии и педиатрии. 2014.
4. Диагноз и прогноз наследственного нефрита у детей с учетом его клинико-морфологического полиморфизма и генетической гетерогенности: Автореферат диссертации/ Цаликова Ф.Д. 1997.
2. Характеристика наследственного нефрита у детей/ Белькевич А.Г., Козыро И.А., Сукало А.В. Актуальные вопросы педиатрии: сборник материалов межрегиональной научно-практической конференции с международным участием, Гродно, 19-20 апреля 2018 г.
3. Наследственные заболевания почек, протекающие с гематурией/ Игнатова М.С., Длин В.В. Российский вестник перинатологии и педиатрии. 2014.
4. Диагноз и прогноз наследственного нефрита у детей с учетом его клинико-морфологического полиморфизма и генетической гетерогенности: Автореферат диссертации/ Цаликова Ф.Д. 1997.