Другие названия и синонимы
Axenfeld-Rieger syndrome.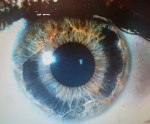
МКБ-10 коды
|
|
Описание
Синдром Аксенфельда-Ригера. Патология, при которой повреждение зрительного органа связано с системными пороками развития. Глазные проявления включают тяжелую дисгенезию угла передней камеры. Системные нарушения включают пороки развития зубов, костей черепа и внутренних органов. Диагноз направлен на проведение гониоскопии, биомикроскопии, глазной тонометрии, компьютерной периметрии и офтальмоскопии. Лечение сводится к инстилляции препаратов для снижения артериального давления из группы ингибиторов сAG, альфа-агонистов, бета-блокаторов и с низкой эффективностью - для выполнения трабекулэктомии.
Дополнительные факты
Синдром Аксенфельда-Ригера - это редкое заболевание, развитие которого обусловлено генетически. Однако только 40% пациентов могут обнаружить хромосомную аномалию и идентифицировать поврежденный ген. Болезнь названа в честь немецкого офтальмолога Т. Аксенфельда и австрийского ученого Г. Ригера. Уровень заболеваемости находится в диапазоне 1: 200 000. Повышение внутриглазного давления, которое проявляется в клинике вторичной глаукомы, происходит в более чем 50% случаев. Патология с одинаковой частотой встречается у мужчин и женщин.
Причины
Этиология заболевания до конца не изучена. Основная теория развития синдрома Аксенфельда-Ригера является генетической. Мутации двух генов, связанных с началом заболевания, не выявлены. Тип наследования является аутосомно-доминантным, но наблюдаются спорадические случаи. Это основание полагать, что существуют другие причины, которые в настоящее время неизвестны.
Патогенез
Доказано, что ген FOXC1 (хромосома 6p25) и PITX2 (хромосома 4q25) кодируют белки, которые действуют как факторы транскрипции и регулируют экспрессию генов. Мутация гена PITX2 нарушает гистогенез глаз, зубов и брюшной полости. FOXC1 отвечает за формирование переднего сегмента глазного яблока. Ученые считают, что изменения в локусе хромосомы 13q14 также играют важную роль в патогенезе, но эта теория еще не подтверждена.
Гистологическое исследование показало, что синдром Аксенфельда-Ригера характеризуется нарушенной миграцией клеток нервного гребня, которые обычно формируются по периферии эмбриональной нервной пластинки даже до закрытия нервной трубки. Это лежит в основе дисгенеза КПК, что может вызвать повышение внутриглазного давления. Гипоплазия экстраокулярных мышц обусловлена нарушением дифференцировки мезодермального комплекса.
Гистологическое исследование показало, что синдром Аксенфельда-Ригера характеризуется нарушенной миграцией клеток нервного гребня, которые обычно формируются по периферии эмбриональной нервной пластинки даже до закрытия нервной трубки. Это лежит в основе дисгенеза КПК, что может вызвать повышение внутриглазного давления. Гипоплазия экстраокулярных мышц обусловлена нарушением дифференцировки мезодермального комплекса.
Клиническая картина
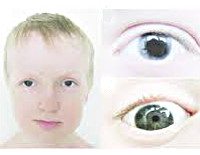
Заболевание двустороннее, но изменения часто бывают асимметричными. Родители в период новорожденности обращают внимание на косметический дефект, который обусловлен аномальной структурой радужной оболочки. Синдром Аксенфельда-Ригера был стабильным в течение длительного времени. Есть опасения по поводу ухудшения зрения, которое происходит после повышения внутриглазного давления.
Черепно-лицевые симптомы включают гипоплазию верхней челюсти, уменьшение размера зубов (микродонтия) или отсутствие отдельных зубов (обычно молоко верхней челюсти или постоянные центральные резцы). Дополнительная кожная складка обнаружена в периумбилической зоне. Часто встречаются нарушения в структуре паха, потеря слуха и умственная отсталость. Гипоспадия может возникнуть у мальчиков.
Черепно-лицевые симптомы включают гипоплазию верхней челюсти, уменьшение размера зубов (микродонтия) или отсутствие отдельных зубов (обычно молоко верхней челюсти или постоянные центральные резцы). Дополнительная кожная складка обнаружена в периумбилической зоне. Часто встречаются нарушения в структуре паха, потеря слуха и умственная отсталость. Гипоспадия может возникнуть у мальчиков.
Возможные осложнения
Синдром Аксенфельда-Ригера в большинстве случаев сопровождается офтальмологической гипертензией. Признаки вторичной глаукомы часто выявляются в подростковом возрасте. Пациенты подвержены риску отслоения сетчатки вследствие пролиферативной витреоретинопатии. Патология часто связана с косоглазием, которое трудно исправить. Наиболее неблагоприятным осложнением синдрома является туберкулез глазного яблока.
Диагностика
Для подтверждения диагноза рекомендуется собрать анамнез, выполнить физическое обследование, осмотреть лицо и полость рта и провести генетическое исследование (прямое секвенирование кодирующей области генов FOXC1 и PITX2). Пренатальное тестирование рекомендуется при наличии синдрома Аксенфельда-Ригера в семье или у родственников по крови. Основные методы диагностики:
Задний эмбриотоксон визуализируется. Пигментные отложения видны вдоль линии Швальбе. Характерно прилипание к структурам КПК процессов, которые простираются от периферии радужки. В отдельных сегментах они достигают линии Швальбе и трабекулярной сети.
• Биомикроскопия глаза. Определяются признаки гипоплазии стромы радужной оболочки, которые более выражены в ресничной зоне. Выявляется истончение листа на поверхности радужки и отек роговицы. Заболевание сопровождается легкой сегментарной гипоплазией. Реже встречаются коктопия и псевдополикория.
• Измерение внутриглазного давления. Измерение ВГД у 50% пациентов показывает признаки офтальмологической гипертонии. При такой патологии очень важно оценивать результаты тонометрии с учетом толщины роговицы. Данные пахиметрии в некоторых случаях указывают на снижение ЦРТ.
• Периметрия компьютеров. При длительном увеличении офтальмотония происходят типичные глаукоматозные изменения в поле зрения. Появляются первые дугообразные скотомы, которые впоследствии становятся множественными. Поле зрения постепенно уменьшается с носовой стороны.
Отклонения оптического диска представлены перипапиллярной атрофией, истончением нейроретинальной талии с височной и нижней сторон. Толщина слоя нервных волокон сетчатки уменьшается в основном в нижнем квадранте.
Дифференциальный диагноз ставится при врожденной глаукоме, аномалии Петерса и гипоплазии радужной оболочки. У пациентов с врожденной глаукомой невозможно увидеть задний эмбриотоксон, что характерно для синдрома Аксенфельда-Ригера. При аномалии Петераса наблюдается центральный дисгенез роговицы и нет признаков гипоплазии радужной оболочки. По мере развития симптомов заболевания указывается консультация офтальмолога и генетика.
Задний эмбриотоксон визуализируется. Пигментные отложения видны вдоль линии Швальбе. Характерно прилипание к структурам КПК процессов, которые простираются от периферии радужки. В отдельных сегментах они достигают линии Швальбе и трабекулярной сети.
• Биомикроскопия глаза. Определяются признаки гипоплазии стромы радужной оболочки, которые более выражены в ресничной зоне. Выявляется истончение листа на поверхности радужки и отек роговицы. Заболевание сопровождается легкой сегментарной гипоплазией. Реже встречаются коктопия и псевдополикория.
• Измерение внутриглазного давления. Измерение ВГД у 50% пациентов показывает признаки офтальмологической гипертонии. При такой патологии очень важно оценивать результаты тонометрии с учетом толщины роговицы. Данные пахиметрии в некоторых случаях указывают на снижение ЦРТ.
• Периметрия компьютеров. При длительном увеличении офтальмотония происходят типичные глаукоматозные изменения в поле зрения. Появляются первые дугообразные скотомы, которые впоследствии становятся множественными. Поле зрения постепенно уменьшается с носовой стороны.
Отклонения оптического диска представлены перипапиллярной атрофией, истончением нейроретинальной талии с височной и нижней сторон. Толщина слоя нервных волокон сетчатки уменьшается в основном в нижнем квадранте.
Дифференциальный диагноз ставится при врожденной глаукоме, аномалии Петерса и гипоплазии радужной оболочки. У пациентов с врожденной глаукомой невозможно увидеть задний эмбриотоксон, что характерно для синдрома Аксенфельда-Ригера. При аномалии Петераса наблюдается центральный дисгенез роговицы и нет признаков гипоплазии радужной оболочки. По мере развития симптомов заболевания указывается консультация офтальмолога и генетика.
Лечение
Лекарства назначаются при повышении внутриглазного давления. Наиболее целесообразно использовать препараты из группы бета-блокаторов, альфа-агонистов и ингибиторов карбоангидразы (CAG), поскольку они помогают снизить выработку внутриглазной жидкости. Следует отметить, что альфа-агонисты следует использовать с осторожностью в детстве из-за возможного подавления функций центральной нервной системы.
Нейропротективная терапия сводится к назначению препаратов, содержащих резвератрол, пирацетам и тиотриазолин. Средняя продолжительность курса 10-14 дней. Эффективность кортиксина была доказана. Эмоксипин вводят парабульбарно. В комплекс лечения должны входить витамины В1, В6. Важно отметить, что целью терапии является предотвращение прогрессирования глаукомных изменений.
Хирургическое вмешательство показано при отсутствии эффекта от консервативной терапии, когда применение комбинированных препаратов не снижает ВГД до контрольных значений. При синдроме Аксенфельда-Ригера трабекулэктомия выполняется с дополнительным использованием антиметаболитов. Если пациент жалуется на светобоязнь с гипоплазией радужной оболочки, рекомендуется иридопластика.
Нейропротективная терапия сводится к назначению препаратов, содержащих резвератрол, пирацетам и тиотриазолин. Средняя продолжительность курса 10-14 дней. Эффективность кортиксина была доказана. Эмоксипин вводят парабульбарно. В комплекс лечения должны входить витамины В1, В6. Важно отметить, что целью терапии является предотвращение прогрессирования глаукомных изменений.
Хирургическое вмешательство показано при отсутствии эффекта от консервативной терапии, когда применение комбинированных препаратов не снижает ВГД до контрольных значений. При синдроме Аксенфельда-Ригера трабекулэктомия выполняется с дополнительным использованием антиметаболитов. Если пациент жалуется на светобоязнь с гипоплазией радужной оболочки, рекомендуется иридопластика.
Лечение синдрома Аксенфельда-Ригера.
Лечение проводится при наличии признаков зрительных нарушений, повышенного офтальмотонуса и светобоязни. В случае чрезмерной чувствительности к свету из-за патологических изменений в радужной оболочке пациенту рекомендуется использовать контактные линзы. Альтернативой является только операция по исправлению дефекта радужной оболочки.|
|
Список литературы
1. The rare Axenfeld-Rieger syndrome with systemic anomalies: A case report and brief review of literature/Wei Song., Xiaodan Hu//Medicine- 2017.
2. A Family with Axenfeld-Rieger Syndrome: Report of the сlinical and Genetic Findings. Yang H.J., Lee Y. K// Korean J Ophthalmol- 2015.
3. Аномалия Ригера с врожденной глаукомой плюс аномалии развития внутренних органов: новая ассоциация или клинический вариант синдрома Аксенфельда-Ригера. (случай из клиники)/Касимов Э. М., Агаева Ф. А. Офтальмология. Научно-практический журнал - 2010 - №3.
4. Genotype-Phenotype сorrelations in Axenfeld-Rieger Malformation and Glaucoma Patients with FOXC1 and PITX2Mutations/ M. Hermina Strungaru; Irina Dinu; Michael A. Walter// Glaucoma - 2007.
2. A Family with Axenfeld-Rieger Syndrome: Report of the сlinical and Genetic Findings. Yang H.J., Lee Y. K// Korean J Ophthalmol- 2015.
3. Аномалия Ригера с врожденной глаукомой плюс аномалии развития внутренних органов: новая ассоциация или клинический вариант синдрома Аксенфельда-Ригера. (случай из клиники)/Касимов Э. М., Агаева Ф. А. Офтальмология. Научно-практический журнал - 2010 - №3.
4. Genotype-Phenotype сorrelations in Axenfeld-Rieger Malformation and Glaucoma Patients with FOXC1 and PITX2Mutations/ M. Hermina Strungaru; Irina Dinu; Michael A. Walter// Glaucoma - 2007.