Другие названия и синонимы
Congenital familial osteopetrosis, Болезнь Албертса-Шенберга, Врожденный остеосклероз, Врожденный семейный диффузный остеосклероз, Гиперостотическая дисплазия, Остеопетроз.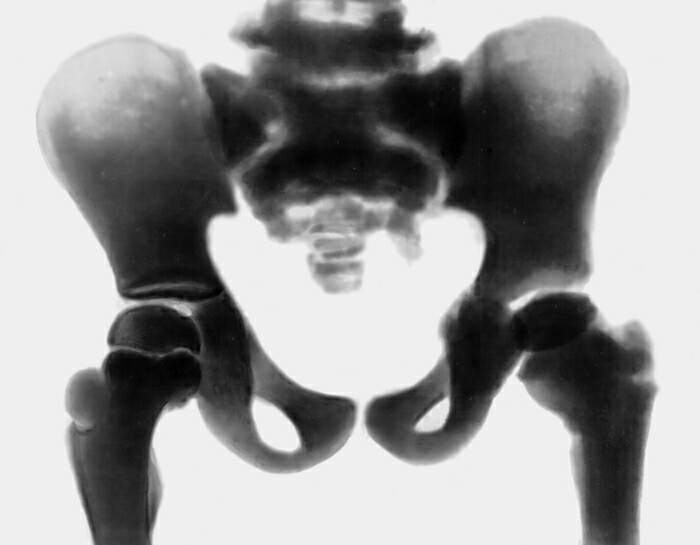
МКБ-10 коды
- МКБ-10
- Q78.2 Остеопетроз
|
|
Описание
Врожденный семейный остеопетроз. Наследственное заболевание с разными типами наследования и клинического течения, которое характеризуется нарушением процессов оссификации костей с их уплотнением и рядом сопутствующих нарушений. Симптомами этого состояния являются ломкость костной ткани (легкое развитие травматических и патологических переломов), проявления тяжелой анемии с гепато- и спленомегалией, в ряде случаев отмечаются неврологические нарушения по причине травматизации нервов, проходящих через костные каналы. Диагностика врожденного семейного остеопетроза основывается на данных рентгенологических исследований, изучения наследственного анамнеза больного, молекулярно-генетических и общих клинических анализов. Специфического лечения этого состояния не существует, в некоторых случаях значительно улучшить состояние больного может трансплантация костного мозга.
Дополнительные факты
Врожденный семейный остеопетроз (мраморная болезнь, синдром Альберс-Шенберга) - группа генетических нарушений со сходными клиническими проявлениями, заключающимся в уплотнении костной ткани с уменьшением диаметра костно-мозговых и других каналов. Одна из форм этого заболевания была впервые описана в 1904-м году немецким хирургом Г. Альберс-Шенбергом, с тех пор наиболее распространенный (аутосомно-доминантный) тип врожденного семейного остеопетроза носит его имя. Это состояние с одинаковой частотой поражает как мужчин, так и женщин, встречаемость зависит от разновидности патологии. Так, наиболее тяжелая по своим клиническим проявлениям аутосомно-рецессивная форма врожденного семейного остеопетроза имеет частоту 1:200 000-300 000, тогда как типы заболевания с аутосомно-доминантным наследованием встречаются у 1-го человека на 20 000 населения. Другое название патологии (мраморная болезнь) обусловлено рентгенологической картиной при ней (зернистость костной ткани, напоминающая мрамор и обусловленная очагами остеопетроза), а также повышенной хрупкостью, но в тоже время твердостью костей. Врожденный семейный остеопетроз является одним из наиболее изученных генетических заболеваний, сопровождающихся гиперостозом.
Причины
Непосредственной причиной всех форм врожденного семейного остеопетроза является нарушение функциональной активности остеокластов - клеток, отвечающих за деструкцию элементов костной ткани. В результате этого нарушается баланс между образованием и разрушением костей, что сопровождается увеличением плотности компактного вещества, уменьшением просвета костномозговых каналов и других отверстий в костях, изменением формы метафизов. Несмотря на остеосклероз, элементы скелета при врожденном семейном остеопетрозе обладают повышенной хрупкостью, что приводит к частым травматическим и патологическим переломам, сколиозам и другим формам деформации позвоночника. Генетическая и молекулярная природа нарушения работы остеокластов неодинакова при различных формах заболевания.
Наиболее часто встречающаяся аутосомно-доминантная форма врожденного семейного остеопетроза (синдром Альберс-Шенберга) обусловлена мутацией гена сLCN7, расположенного на 16-й хромосоме. Он кодирует последовательности одной из субъединиц хлор-специфичного ионного канала, который особенно распространен на поверхности мембран остеокластов. Он участвует в процессе образования соляной кислоты, которая необходима для растворения солей кальция, входящих в состав костной ткани. В результате дефекта гена сLCN7 структура ионного канала изменяется и приводит к нарушению его функции, в результате чего выделение соляной кислоты сильно снижается, что и является причиной врожденного семейного остеопетроза.
Врожденный семейный остеопетроз с аутосомно-рецессивным типом наследования представляет собой более генетически гетерогенное состояние, оно может быть обусловлено мутациями нескольких генов. Классический тип этой патологии, по данным современной генетики, обусловлен дефектом гена TCIRG1, локализованного на 11-й хромосоме. Продуктом его экспрессии является протеин под названием «Т-клеточного иммунного регулятора», и он же представляет собой одну из субъединиц трансмембранного белка вакуолей остеокластов, проявляющего свойства АТФ-зависимого протонного насоса. Наряду с рядом хлорных каналов, он участвует в образовании соляной кислоты, которая, как и при аутосомно-доминантном варианте врожденного семейного остеопетроза необходима для деструкции костной ткани и функционирования остеокластов. Мутации гена TCIRG1 обуславливают примерно половину всех случаев данной формы заболевания.
Другие генетические дефекты намного реже приводят к развитию врожденного семейного остеопетроза с аутосомно-рецессивным типом наследования. Так, примерно у 10% таких больных выявляются мутации гена сLCN7, ассоциированного с доминантной формой патологии - но при этом характер генетического дефекта более выраженный и приводит к прекращению экспрессии белков хлор-специфичного ионного канала. Еще более редкий тип врожденного семейного остеопетроза вызывается мутацией гена OSTM1, расположенного на 6-й хромосоме - он кодирует одноименный протеин, участвующий во внутриклеточной передаче информации в остеокластах. В результате данного генетического дефекта нарушаются практически все функции этих клеток, даже при сохраненной способности синтезировать соляную кислоту. По некоторым данным, существует разновидность врожденного семейного остеопетроза, которая передается по механизму сцепленного с полом - врачи-генетики связывают ее с мутациями гена IBKG.
Наиболее часто встречающаяся аутосомно-доминантная форма врожденного семейного остеопетроза (синдром Альберс-Шенберга) обусловлена мутацией гена сLCN7, расположенного на 16-й хромосоме. Он кодирует последовательности одной из субъединиц хлор-специфичного ионного канала, который особенно распространен на поверхности мембран остеокластов. Он участвует в процессе образования соляной кислоты, которая необходима для растворения солей кальция, входящих в состав костной ткани. В результате дефекта гена сLCN7 структура ионного канала изменяется и приводит к нарушению его функции, в результате чего выделение соляной кислоты сильно снижается, что и является причиной врожденного семейного остеопетроза.
Врожденный семейный остеопетроз с аутосомно-рецессивным типом наследования представляет собой более генетически гетерогенное состояние, оно может быть обусловлено мутациями нескольких генов. Классический тип этой патологии, по данным современной генетики, обусловлен дефектом гена TCIRG1, локализованного на 11-й хромосоме. Продуктом его экспрессии является протеин под названием «Т-клеточного иммунного регулятора», и он же представляет собой одну из субъединиц трансмембранного белка вакуолей остеокластов, проявляющего свойства АТФ-зависимого протонного насоса. Наряду с рядом хлорных каналов, он участвует в образовании соляной кислоты, которая, как и при аутосомно-доминантном варианте врожденного семейного остеопетроза необходима для деструкции костной ткани и функционирования остеокластов. Мутации гена TCIRG1 обуславливают примерно половину всех случаев данной формы заболевания.
Другие генетические дефекты намного реже приводят к развитию врожденного семейного остеопетроза с аутосомно-рецессивным типом наследования. Так, примерно у 10% таких больных выявляются мутации гена сLCN7, ассоциированного с доминантной формой патологии - но при этом характер генетического дефекта более выраженный и приводит к прекращению экспрессии белков хлор-специфичного ионного канала. Еще более редкий тип врожденного семейного остеопетроза вызывается мутацией гена OSTM1, расположенного на 6-й хромосоме - он кодирует одноименный протеин, участвующий во внутриклеточной передаче информации в остеокластах. В результате данного генетического дефекта нарушаются практически все функции этих клеток, даже при сохраненной способности синтезировать соляную кислоту. По некоторым данным, существует разновидность врожденного семейного остеопетроза, которая передается по механизму сцепленного с полом - врачи-генетики связывают ее с мутациями гена IBKG.
Клиническая картина
Выраженность симптомов врожденного семейного остеопетроза и тяжесть его течения находится в сильной зависимости от формы этого заболевания. Наиболее ранними и явными проявлениями обладают аутосомно-рецессивные типы этой патологии, которые проявляются сразу при рождении, а в некоторых случаях и внутриутробно на заключительных этапах вынашивания ребенка. Ведущим проявлением на этом этапе развития врожденного семейного остеопетроза является тяжелейшая анемия, вызванная дефицитом красного костного мозга из-за сужения костномозговых каналов. Она приводит к многочисленным вторичным нарушениям - гепато- и спленомегалии, имеющие прогрессирующий характер, бледность кожных покровов, признаки кислородного голодания тканей. Нередко у страдающих врожденным семейным остеопетрозом младенцев определяют гидроцефалию, в дальнейшем многие больные теряют слух и зрение из-за сужения отверстий черепа, через которые проходят соответствующие нервы. Лицо таких больных имеет характерные особенности - широко расставленные глаза, вдавленная переносица, толстые и диспропорциональные губы. Многочисленные нарушения при аутосомно-рецессивной форме врожденного семейного остеопетроза имеют тенденцию к прогрессированию, что нередко приводит к смерти больных до достижения ими 3-8 лет.
Диагностика
Для диагностики врожденного семейного остеопетроза применяют рентгенологические методики, общие анализы крови, молекулярно-генетические исследования, изучение наследственного анамнеза больного. На рентгенограммах выявляют общее уплотнение костной ткани (диффузный остеосклероз), выраженность которого больше при аутосомно-рецессивных вариантах заболевания. В некоторых элементах скелета (фаланги пальцев, подвздошные кости, позвонки) остеопетроз имеет очаговый характер, создавая характерную рентгенологическую картину, известную как «кость в кости». В длинных трубчатых костях изменяется форма метафизов и размеры костномозгового канала - от незначительного уменьшения при доброкачественном типе врожденного семейного остеопетроза до практически полного исчезновения при рецессивном. На рентгенограммах также могут выявляться признаки гидроцефалии, искривления позвоночника, у взрослых - следы многочисленных заживших переломов.
В анализах крови определяется резкое снижение уровня эритроцитов и гемоглобина, нередко снижаются показатели относительно других клеточных элементов. У детей с рецессивной формой врожденного семейного остеопетроза это может иметь жизнеугрожающий характер, тогда как у взрослых (аутосомно-доминантная форма) анемия выражена слабо или вообще не обнаруживается. Молекулярно-генетические исследования наиболее часто сводятся к прямому секвенированию гена TCIRG1, мутации которого приводят к злокачественному типу этого заболевания. Поводом для такого анализа служат, помимо характерной клинической картины, наличие подобных проявлений у родственников родителей больного - это может указывать на аутосомно-рецессивный характер передачи болезни.
В анализах крови определяется резкое снижение уровня эритроцитов и гемоглобина, нередко снижаются показатели относительно других клеточных элементов. У детей с рецессивной формой врожденного семейного остеопетроза это может иметь жизнеугрожающий характер, тогда как у взрослых (аутосомно-доминантная форма) анемия выражена слабо или вообще не обнаруживается. Молекулярно-генетические исследования наиболее часто сводятся к прямому секвенированию гена TCIRG1, мутации которого приводят к злокачественному типу этого заболевания. Поводом для такого анализа служат, помимо характерной клинической картины, наличие подобных проявлений у родственников родителей больного - это может указывать на аутосомно-рецессивный характер передачи болезни.
|
|
Лечение
Специфическое лечение аутосомно-рецессивных форм врожденного семейного остеопетроза (как и доминантных) не разработано - некоторые исследователи в качестве такового указывают на трансплантацию костного мозга. Так как некоторые остеокласты являются вариантом тканевых макрофагов (то есть, имеют костномозговое происхождение) такой метод лечения способен улучшить состояние больного за счет «замены» собственных клеток донорскими остеокластами без генетических дефектов. Действительно, в ряде случаев врожденного семейного остеопетроза трансплантация костного мозга приводила к улучшению, но примеров полного выздоровления зафиксировано не было. Остальные терапевтические мероприятия имеют поддерживающий и симптоматический характер - применение антианемических средств (препаратов железа, эритропоэтинов), витамина Д, лечебная физкультура и гимнастика. При аутосомно-доминантной форме врожденного семейного остеопетроза заметное улучшение наблюдается при санаторно-курортном лечении.