Другие названия и синонимы
Pearson 's syndrome.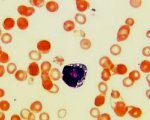
Описание
Синдром Пирсона. Это мультисистемное митохондриальное заболевание, при котором наблюдаются нарушения кроветворения, недостаточность функций поджелудочной железы и печени. Патология развивается на фоне дупликации (удвоения) или спорадической делеции (потери одного сайта) материнской мДНК. Синдром возникает в детстве при тяжелой анемии, длительной диарее и задержке психомоторного развития. Диагностика заболевания включает гематологический и биохимический анализы крови, миелограмму, генетические тесты. Поддерживающая терапия: заместительная ферментная терапия, переливание компонентов крови, метаболических агентов.
|
|
Дополнительные факты
Синдром назван в честь профессора Ховарда Пирсона, который описал его клинические симптомы в 1979 году. 10 лет спустя была установлена связь между патологией и молекулярно-генетическими дефектами в наследственном материале митохондрий. Раньше в мире было не более 100 случаев синдрома Пирсона, но по мере улучшения диагностических критериев была установлена истинная частота заболевания - 1: 5000-1: 10 000 новорожденных. Синдром характеризуется тяжелым неблагоприятным течением, отсутствием этиопатогенетической терапии.
Причины
Синдром Пирсона возникает при дублировании или удалении фрагмента митохондриальной ДНК. Эта мутация передается только от матери, так как отцовские митохондрии не попадают в яйцеклетку при зачатии или разрушаются сразу после проникновения. Специалисты-генетики еще не смогли установить закономерность развития и триггерные факторы заболевания, поэтому оно считается спорадическим.
Патогенез
Митохондрии - это органеллы, которые находятся во всех клетках тела (кроме зрелых эритроцитов) и производят до 95% энергии. Энергия в виде молекул АТФ используется для поддержания жизненно важных процессов. Каждая клетка содержит несколько тысяч митохондрий, которые содержат 2-10 кольцевых молекул ДНК, которые могут реплицироваться, транскрибироваться и транслироваться независимо от основной генетической информации в ядре клетки.
Когда митохондриальная ДНК мутирует, функция этих органелл снижается, затрагивая все органы и ткани, поэтому синдром является органо-неспецифическим и имеет чрезвычайно тяжелые проявления. У пациентов Пирсона нарушены электронный транспорт и окислительное фосфорилирование - важные шаги в образовании АТФ. В первую очередь повреждаются самые изменчивые ткани: нервная система, мышцы, в том числе миокард, печень и поджелудочная железа.
Когда митохондриальная ДНК мутирует, функция этих органелл снижается, затрагивая все органы и ткани, поэтому синдром является органо-неспецифическим и имеет чрезвычайно тяжелые проявления. У пациентов Пирсона нарушены электронный транспорт и окислительное фосфорилирование - важные шаги в образовании АТФ. В первую очередь повреждаются самые изменчивые ткани: нервная система, мышцы, в том числе миокард, печень и поджелудочная железа.
Клиническая картина
В большинстве случаев синдром начинается с сидеробластной анемии в первые месяцы жизни пациента. У ребенка бледная кожа с голубоватым или зеленоватым оттенком, вялость, постоянная сонливость. Ребенок плохо сосет, ест мало молока и медленно набирает вес. Анемию нельзя лечить препаратами железа, витамином в12 или фолиевой кислотой.
Второй типичный симптом синдрома Пирсона - хроническая диарея, возникающая из-за недостаточности поджелудочной железы. Частота кала у больных детей достигает 10-20 раз в сутки, кал жидкий, зловонный, обычно отмечаются комочки простокваши. Характеризуется частой срыгиванием, сильными коликами, периодическими приступами рвоты, вызванными метаболическими кризами.
Синдром может дополняться неврологическими расстройствами. Поражение нервно-мышечной системы проявляется стойкой гипотонией, атаксией, задержкой формирования крупной и мелкой моторики. Иногда у пациентов развивается подострая некротическая энцефалопатия, которая клинически напоминает синдром Ли и сопровождается тяжелой умственной отсталостью.
При «легком» варианте болезни Пирсона с возрастом его можно трансформировать в другое заболевание: синдром Кернса-Стейса. У этих детей задержка роста, задержка физического развития и нарушения координации, такие как атаксия. Повреждение глаза представлено пигментной ретинопатией, офтальмоплегией. Характерны генерализованные миопатии, поражение миокарда с появлением аритмий.
Ассоциированные симптомы: Диарея у ребенка. Нейтропения. Понос (диарея).
Второй типичный симптом синдрома Пирсона - хроническая диарея, возникающая из-за недостаточности поджелудочной железы. Частота кала у больных детей достигает 10-20 раз в сутки, кал жидкий, зловонный, обычно отмечаются комочки простокваши. Характеризуется частой срыгиванием, сильными коликами, периодическими приступами рвоты, вызванными метаболическими кризами.
Синдром может дополняться неврологическими расстройствами. Поражение нервно-мышечной системы проявляется стойкой гипотонией, атаксией, задержкой формирования крупной и мелкой моторики. Иногда у пациентов развивается подострая некротическая энцефалопатия, которая клинически напоминает синдром Ли и сопровождается тяжелой умственной отсталостью.
При «легком» варианте болезни Пирсона с возрастом его можно трансформировать в другое заболевание: синдром Кернса-Стейса. У этих детей задержка роста, задержка физического развития и нарушения координации, такие как атаксия. Повреждение глаза представлено пигментной ретинопатией, офтальмоплегией. Характерны генерализованные миопатии, поражение миокарда с появлением аритмий.
Ассоциированные симптомы: Диарея у ребенка. Нейтропения. Понос (диарея).
Возможные осложнения
Со временем болезнь прогрессирует с вовлечением других систем организма. При поражении почек выявляются тубулопатии, синдром Тони-Дебре-Фанкони. Патология печени характеризуется жировым гепатозом, гепатомегалией, цитолизом. Фиброзные изменения поджелудочной железы вызывают снижение синтеза инсулина, что проявляется инсулинозависимым сахарным диабетом.
Наиболее опасным осложнением у детей с болезнью Пирсона является лактоацидоз, при котором постепенно нарушается сократительная способность сердца, усиливаются неврологические симптомы и обостряются желудочно-кишечные расстройства. В дальнейшем возникает коллапс, переохлаждение, острая почечная недостаточность. Лактоацидоз заканчивается глубокой комой. Смерть наступает у 60-70% пациентов даже во время реанимации.
Наиболее опасным осложнением у детей с болезнью Пирсона является лактоацидоз, при котором постепенно нарушается сократительная способность сердца, усиливаются неврологические симптомы и обостряются желудочно-кишечные расстройства. В дальнейшем возникает коллапс, переохлаждение, острая почечная недостаточность. Лактоацидоз заканчивается глубокой комой. Смерть наступает у 60-70% пациентов даже во время реанимации.
Диагностика
При осмотре обращают внимание на истощение, бледность кожных покровов, задержку психомоторного развития. Клинические симптомы аналогичны симптомам других митохондриальных заболеваний, что требует высокого профессионализма педиатра и обширных диагностических мероприятий. Для верификации диагноза синдрома Пирсона назначаются следующие лабораторные и инструментальные исследования:
• Гематологические тесты. В анализе крови выявляются признаки макроцитарной анемии, ретикулоцитопении, в тяжелых случаях наблюдаются тромбоцитопения и нейтропения. При изучении показателей обмена железа типичным проявлением является повышение уровня ферритина при нормальном количестве железа в крови.
• Биохимические исследования. В сыворотке крови снижается концентрация амилазы поджелудочной железы, как правило, развивается синдром цитолиза с повышением АЛТ, АСТ и щелочной фосфатазы. На лактоацидоз указывает резкое повышение уровня молочной кислоты в крови, снижение уровня бикарбоната и падение pH ниже 7,3.
• Биопсия костного мозга. Миелограмма нужна для дифференциации различных вариантов нарушений кроветворения. Болезнь Пирсона характеризуется вакуолизацией цитоплазмы предшественников эритроцитов, наличием кольцевых сидеробластов после окрашивания берлинской синей.
• Молекулярно-генетические исследования. Для подтверждения диагноза из митохондрий лейкоцитов извлекается ДНК с последующим ее генетическим тестированием. В этом случае определяется большая делеция или дупликация мДНК, часто наблюдаются признаки гетероплазмии.
С учетом основных клинических проявлений дополнительно используются методы инструментальной визуализации. Ультрасонография органов брюшной полости выявляет гепатомегалию, увеличение и диффузные изменения поджелудочной железы. При неврологической симптоматике показана КТ головного мозга, подтверждающая очаговые атрофические поражения. Для исключения сопутствующих врожденных пороков развития проводится эхокардиография и рентгенография грудной клетки.
• Гематологические тесты. В анализе крови выявляются признаки макроцитарной анемии, ретикулоцитопении, в тяжелых случаях наблюдаются тромбоцитопения и нейтропения. При изучении показателей обмена железа типичным проявлением является повышение уровня ферритина при нормальном количестве железа в крови.
• Биохимические исследования. В сыворотке крови снижается концентрация амилазы поджелудочной железы, как правило, развивается синдром цитолиза с повышением АЛТ, АСТ и щелочной фосфатазы. На лактоацидоз указывает резкое повышение уровня молочной кислоты в крови, снижение уровня бикарбоната и падение pH ниже 7,3.
• Биопсия костного мозга. Миелограмма нужна для дифференциации различных вариантов нарушений кроветворения. Болезнь Пирсона характеризуется вакуолизацией цитоплазмы предшественников эритроцитов, наличием кольцевых сидеробластов после окрашивания берлинской синей.
• Молекулярно-генетические исследования. Для подтверждения диагноза из митохондрий лейкоцитов извлекается ДНК с последующим ее генетическим тестированием. В этом случае определяется большая делеция или дупликация мДНК, часто наблюдаются признаки гетероплазмии.
С учетом основных клинических проявлений дополнительно используются методы инструментальной визуализации. Ультрасонография органов брюшной полости выявляет гепатомегалию, увеличение и диффузные изменения поджелудочной железы. При неврологической симптоматике показана КТ головного мозга, подтверждающая очаговые атрофические поражения. Для исключения сопутствующих врожденных пороков развития проводится эхокардиография и рентгенография грудной клетки.
Лечение
Помощь пациентам с синдромом Пирсона требует мультисистемного подхода с участием педиатров, детских гематологов, гастроэнтерологов; по показаниям к терапии привлекаются генетики, неврологи и нефрологи. Пациентам требуется поддерживающая терапия на протяжении всей жизни, в том числе:
• Переливание компонентов крови. Только так можно исправить тяжелую анемию, восполнить недостаток тромбоцитов для улучшения свертывания крови, повысить уровень лейкоцитов для активации иммунной системы.
• Стимуляция гранулоцитопоэза. При критическом снижении количества нейтрофилов применяют препараты гранулоцитарного колониестимулирующего фактора, усиливающие образование лейкоцитов в костном мозге.
• Ферментная терапия. Препараты ферментов поджелудочной железы и желчных кислот используются для улучшения переваривания пищи в тонком кишечнике, уменьшения признаков плохого пищеварения и нормализации стула.
• Метаболические препараты. Чтобы стимулировать перенос электронов в дыхательной цепи митохондрий, активировать выработку АТФ, рекомендуется принимать кофермент Q10, левокарнитин. Обычно назначают витамины В2, РР, Е.
• Коррекция pH крови. С целью устранения лактоацидоза вводят растворы бикарбоната натрия, метиленового синего, трисамина. В тяжелых случаях лечение дополняется гемодиализом или перитонеальным диализом с безлактатным диализатом.
В Израиле начались клинические испытания использования клеточной терапии при синдроме Пирсона. Эксперимент предполагает пересадку стволовых клеток со здоровыми митохондриями в организм ребенка. Исследователи ожидают улучшения качества жизни пациентов, уменьшения потребности в переливаниях крови и уменьшения потребления ферментов. В случае успеха этот метод станет ключом к этиопатогенетическому лечению других митохондриальных патологий.
• Переливание компонентов крови. Только так можно исправить тяжелую анемию, восполнить недостаток тромбоцитов для улучшения свертывания крови, повысить уровень лейкоцитов для активации иммунной системы.
• Стимуляция гранулоцитопоэза. При критическом снижении количества нейтрофилов применяют препараты гранулоцитарного колониестимулирующего фактора, усиливающие образование лейкоцитов в костном мозге.
• Ферментная терапия. Препараты ферментов поджелудочной железы и желчных кислот используются для улучшения переваривания пищи в тонком кишечнике, уменьшения признаков плохого пищеварения и нормализации стула.
• Метаболические препараты. Чтобы стимулировать перенос электронов в дыхательной цепи митохондрий, активировать выработку АТФ, рекомендуется принимать кофермент Q10, левокарнитин. Обычно назначают витамины В2, РР, Е.
• Коррекция pH крови. С целью устранения лактоацидоза вводят растворы бикарбоната натрия, метиленового синего, трисамина. В тяжелых случаях лечение дополняется гемодиализом или перитонеальным диализом с безлактатным диализатом.
В Израиле начались клинические испытания использования клеточной терапии при синдроме Пирсона. Эксперимент предполагает пересадку стволовых клеток со здоровыми митохондриями в организм ребенка. Исследователи ожидают улучшения качества жизни пациентов, уменьшения потребности в переливаниях крови и уменьшения потребления ферментов. В случае успеха этот метод станет ключом к этиопатогенетическому лечению других митохондриальных патологий.
|
|
Прогноз
Продолжительность жизни пациентов, у которых проявление заболевания произошло в течение первых месяцев после рождения, не превышает 2-3 лет. Несмотря на достижения современной медицины, прогноз остается неблагоприятным, большинство смертей происходит из-за усиления лактоацидоза. Учитывая спорадический характер заболевания, генетическое консультирование пары и пренатальная диагностика неэффективны в качестве профилактических мер.
Список литературы
1. Случай синдрома Пирсона у грудного ребенка. Клиническое наблюдение/ А.С. Панченко, О.Г. Максимова, Е.П. Мацеха, О.И. Кряжева// Педиатрия. 2018. №11.
2. Митохондриальная патология у детей/ В.М. Студеникин, О.В. Глоба// Лечащий врач. 2016. №1.
3. Синдром Пирсона у ребенка раннего возраста/ Е.Б. Копилова, О.И. Вотякова, Е.Г. Кузнецова, И.В. Князева и // Лечение и профилактика. Научно-практический медицинский журнал. 2016. №1(17).
4. Синдром Пирсона/ Г.С. Овсянникова, И.И. Калинина, Д.Д. Байдильдина, Л.А. Хачатрян// Журнал «Педиатрия» им. Г.Н. Сперанского. 2014. №6.
2. Митохондриальная патология у детей/ В.М. Студеникин, О.В. Глоба// Лечащий врач. 2016. №1.
3. Синдром Пирсона у ребенка раннего возраста/ Е.Б. Копилова, О.И. Вотякова, Е.Г. Кузнецова, И.В. Князева и // Лечение и профилактика. Научно-практический медицинский журнал. 2016. №1(17).
4. Синдром Пирсона/ Г.С. Овсянникова, И.И. Калинина, Д.Д. Байдильдина, Л.А. Хачатрян// Журнал «Педиатрия» им. Г.Н. Сперанского. 2014. №6.