Другие названия и синонимы
Holt-Oram syndrome, Предсердно-пальцевая дисплазия, Синдром «рука-сердце».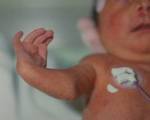
МКБ-10 коды
|
|
Описание
Синдром Холта-Орама. Это редкая наследственная патология, характеризующаяся морфологическими аномалиями верхних конечностей, различными врожденными пороками сердца. Возникает из-за мутации гена TBX5, наследуется по аутосомно-доминантному типу. Для диагностики синдрома Холта-Орама назначают рентген руки, эхокардиографию и электрокардиографию, наличие мутации подтверждается генетическим тестированием. В курс лечения входят операции по устранению пороков сердца, помощь травматологов-ортопедов при тяжелых деформациях опорно-двигательного аппарата.
Дополнительные факты
Синдром Холта-Орама имеет дополнительные названия в медицинской литературе: синдром руки-сердца, атрио-цифровая дисплазия. Типичные клинические проявления болезни впервые были описаны в 1960 году британскими кардиологами Мэри Холт и Сэмюэлем Орамом, от которых болезнь получила свое название. Точные генетические причины развития аномалий скелета и сердца были установлены в 1997 году. Болезнь Холта-Орама относится к орфанным патологиям, встречается с частотой 1 случай на 100 тысяч населения, не имеет половых различий.
Причины
Заболевание вызвано мутациями в гене TBX5, который расположен на длинном плече хромосомы 12 в локусе 12q24.21. Патология имеет аутосомно-доминантный тип наследования, однако нередки случаи спонтанного возникновения мутации у ребенка, имеющего полностью здоровые родители. Мутация гена имеет разную степень выраженности, что определяет вариабельность клинической картины синдрома.
Патогенез
Типичные морфологические изменения при болезни Холта-Орама связаны с отсутствием или недостаточным синтезом фактора транскрипции Tbx5, который принадлежит к семейству T-боксов. Он регулирует многие процессы образования соединительной ткани во время внутриутробного развития, включая развитие предсердной и межжелудочковой перегородок, проводящей системы сердца, мышц и сухожилий.
При недостаточном уровне белка Tbx5 обычно нарушается структура перегородки сердца, аортального клапана, именно в этих зонах возникает до 95% всех врожденных дефектов у страдающих синдромом Холта-Орама. Также наличие фактора транскрипции является обязательным условием дифференцировки кардиомиоцитов проводящей системы сердца, поэтому патология часто сопровождается нарушением ритма.
При недостаточном уровне белка Tbx5 обычно нарушается структура перегородки сердца, аортального клапана, именно в этих зонах возникает до 95% всех врожденных дефектов у страдающих синдромом Холта-Орама. Также наличие фактора транскрипции является обязательным условием дифференцировки кардиомиоцитов проводящей системы сердца, поэтому патология часто сопровождается нарушением ритма.
Клиническая картина
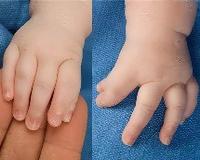
Основной симптом синдрома Холта-Орама - аномалии строения верхней конечности, которые выявляются у 100% пациентов. В основном они проявляются изолированной гипоплазией тенара - ладонным возвышением у основания большого пальца, появлением третьей фаланги на большом пальце, лево-правой асимметрией. В тяжелых случаях синдрома возникают тяжелые пороки, приводящие к инвалидности: фокомелия (конечности, похожие на тюленя).
Второй по частоте симптом, наблюдаемый у 85% пациентов, - это врожденные пороки сердца. Чаще всего у пациентов наблюдается дефект межпредсердной перегородки (44,4%), дефект межжелудочковой перегородки (29,4%). Гораздо реже встречаются грубые сердечные аномалии: тетрада Фалло, синдром гипоплазии левых отделов сердца, коарктация аорты.
Третье по частоте проявление синдрома Холта-Орама - нарушения ритма и проводимости, которые регистрируются в 40% случаев. Они представлены синусовой брадикардией, блокадой правой ножки пучка Гиса, атриовентрикулярной диссоциацией или синдромом слабости синусового узла. В редких случаях определяются признаки эмбриогенеза: гипертелоризм, волчья пасть, отсутствие большой грудной мышцы.
Второй по частоте симптом, наблюдаемый у 85% пациентов, - это врожденные пороки сердца. Чаще всего у пациентов наблюдается дефект межпредсердной перегородки (44,4%), дефект межжелудочковой перегородки (29,4%). Гораздо реже встречаются грубые сердечные аномалии: тетрада Фалло, синдром гипоплазии левых отделов сердца, коарктация аорты.
Третье по частоте проявление синдрома Холта-Орама - нарушения ритма и проводимости, которые регистрируются в 40% случаев. Они представлены синусовой брадикардией, блокадой правой ножки пучка Гиса, атриовентрикулярной диссоциацией или синдромом слабости синусового узла. В редких случаях определяются признаки эмбриогенеза: гипертелоризм, волчья пасть, отсутствие большой грудной мышцы.
Возможные осложнения
Пациенты с синдромом Холта-Орама часто отстают в физическом росте и развитии с раннего детства, тогда как умственное развитие происходит в зависимости от возраста. Если не проводить операцию на сердце, наблюдаются нарушения кровообращения на фоне пороков сердца, вплоть до тяжелой сердечной недостаточности. При длительном существовании синдрома клеточный иммунитет снижается из-за частых ОРЗ, пневмонии, среднего отита и синусита.
Диагностика
Поскольку первые признаки синдрома заметны уже в раннем детстве, ребенка осматривает педиатр; по показаниям к диагностике привлекаются детский кардиохирург, генетик, ортопед-травматолог. Заподозрить диагноз можно по наличию специфических деформаций верхних конечностей. Для подтверждения синдрома Холта-Орама назначают следующие методы диагностики:
• Рентгенография руки. Рентген показывает гипоплазию тенара, появление третьей фаланги на большом пальце одной или обеих рук и недостаточную оппозицию большого пальца. Изредка выявляются более серьезные деформации костей кисти с вовлечением в процесс пояса верхней конечности.
• Эхокардиография. Ультразвуковое исследование сердца - ведущий метод диагностики врожденных аномалий, характерных для синдрома. При осмотре можно визуализировать структурные изменения органа, оценить его функциональные возможности. Для уточнения диагноза используется рентгенография ОГК.
• Электрокардиография. Метод позволяет на ранней стадии выявить признаки синусовых аритмий периодического и апериодического типа, синоатриальной блокады, синусовой тахикардии, предсердной экстрасистолии и других типичных нарушений ритма. В некоторых случаях для более точной диагностики рекомендуется ежедневный мониторинг ЭКГ.
• Генетический тест. Поиск конкретной мутации путем секвенирования экзонов - основной метод подтверждения диагноза, который используется в сомнительных случаях в научных исследованиях. В повседневной практике, как правило, достаточно стандартных клинических и инструментальных методов.
• Рентгенография руки. Рентген показывает гипоплазию тенара, появление третьей фаланги на большом пальце одной или обеих рук и недостаточную оппозицию большого пальца. Изредка выявляются более серьезные деформации костей кисти с вовлечением в процесс пояса верхней конечности.
• Эхокардиография. Ультразвуковое исследование сердца - ведущий метод диагностики врожденных аномалий, характерных для синдрома. При осмотре можно визуализировать структурные изменения органа, оценить его функциональные возможности. Для уточнения диагноза используется рентгенография ОГК.
• Электрокардиография. Метод позволяет на ранней стадии выявить признаки синусовых аритмий периодического и апериодического типа, синоатриальной блокады, синусовой тахикардии, предсердной экстрасистолии и других типичных нарушений ритма. В некоторых случаях для более точной диагностики рекомендуется ежедневный мониторинг ЭКГ.
• Генетический тест. Поиск конкретной мутации путем секвенирования экзонов - основной метод подтверждения диагноза, который используется в сомнительных случаях в научных исследованиях. В повседневной практике, как правило, достаточно стандартных клинических и инструментальных методов.
Лечение
Лечебные мероприятия подбираются с учетом характера и степени клинических проявлений синдрома. В основе лечения лежит помощь кардиохирургов по устранению врожденных пороков сердца, что позволяет устранить нарушения кровообращения, обеспечить пациентам высокую продолжительность жизни и уровень жизни. В зависимости от типа дефекта хирургическое вмешательство проводится в разном возрасте:
• При дефекте межжелудочковой перегородки оптимальный возраст коррекции от 1 до 2-2,5 лет, что позволяет избежать серьезных осложнений и обеспечить нормальное физическое развитие ребенка.
• В случае дефекта межпредсердной перегородки наилучшие результаты коррекции достигаются при проведении операции в дошкольном возрасте (3-6 лет).
• При тяжелых пороках (например, тетрада Фалло) кардиохирургия проводится в 4-6 месяцев, а при наличии приступов одышки и цианоза - еще раньше.
• При угрожающих жизни аритмиях показана имплантация кардиостимулятора, если медикаментозное лечение неэффективно.
Помимо кардиохирургии, пациенту может потребоваться реконструктивная операция для уменьшения степени дефекта верхней конечности, повышения ее функциональности. На протяжении всей жизни пациент с болезнью Холта-Орама находится на амбулаторном лечении у кардиолога, ортопеда и генетика.
• При дефекте межжелудочковой перегородки оптимальный возраст коррекции от 1 до 2-2,5 лет, что позволяет избежать серьезных осложнений и обеспечить нормальное физическое развитие ребенка.
• В случае дефекта межпредсердной перегородки наилучшие результаты коррекции достигаются при проведении операции в дошкольном возрасте (3-6 лет).
• При тяжелых пороках (например, тетрада Фалло) кардиохирургия проводится в 4-6 месяцев, а при наличии приступов одышки и цианоза - еще раньше.
• При угрожающих жизни аритмиях показана имплантация кардиостимулятора, если медикаментозное лечение неэффективно.
Помимо кардиохирургии, пациенту может потребоваться реконструктивная операция для уменьшения степени дефекта верхней конечности, повышения ее функциональности. На протяжении всей жизни пациент с болезнью Холта-Орама находится на амбулаторном лечении у кардиолога, ортопеда и генетика.
Прогноз
Прогноз определяется тяжестью поражения сердца: незначительные дефекты можно полностью устранить с помощью операции на сердце, но серьезные аномалии сердца с высоким риском смерти встречаются редко. Из-за спонтанного характера мутации мер первичной профилактики синдрома не разработано. Вторичная профилактика включает своевременную диагностику и операцию на сердце.
Список литературы
1. Множественные мальформации сердца у пациента с синдромом Холта-Орама/ И.А. Сойнов, Д.А. Дульцева, А.В. Лейкхеман, А.Н. Архипов// Российский вестник перинатологии и педиатрии. 2020. №5.
2. Клинический случай: синдром Холта-Орама/ А.С. Хайбуллина // 50-я ежегодная научно-практическая конференция студентов и молодых учёных по итогам летней производственной практики. 2017.
3. Синдром Холта-Орама/ О.Л. Цимбалиста, Н.Н. Фоменко, Т.М. Мельник, Е.Д. Шустакевич// Здоровье ребенка. 2010. №6.
4. Клинический случай синдрома Холта-Орама у ребенка / С.Я. Волгина, Н.И. Клейменова // Казанский медицинский журнал. 2009. №6.
2. Клинический случай: синдром Холта-Орама/ А.С. Хайбуллина // 50-я ежегодная научно-практическая конференция студентов и молодых учёных по итогам летней производственной практики. 2017.
3. Синдром Холта-Орама/ О.Л. Цимбалиста, Н.Н. Фоменко, Т.М. Мельник, Е.Д. Шустакевич// Здоровье ребенка. 2010. №6.
4. Клинический случай синдрома Холта-Орама у ребенка / С.Я. Волгина, Н.И. Клейменова // Казанский медицинский журнал. 2009. №6.
|
|