Другие названия и синонимы
Kabuki syndrome, Синдром грима Кабуки, Синдром маски Кабуки, Синдром Ниикава-Куроки.
МКБ-10 коды
|
|
Описание
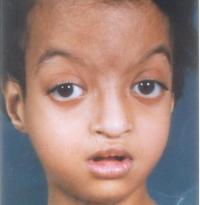
Это редкое моногенное заболевание, характеризующееся специфическими фенотипическими признаками и интеллектуальным дефицитом. Пациенты имеют черты лица (миндалевидные глаза, косоглазие, дугообразные брови, широкая переносица, низко посаженные выступающие уши ), Скелетные аномалии, дефекты внутренних органов, олигофрения. Диагноз основывается на клинических критериях, подтвержденных генетическими тестами. Лечение сводится к коррекции врожденных аномалий, угрожающих жизни и здоровью ребенка, лечению сопутствующих расстройств (аутоиммунных, эндокринных, инфекционных и ;).
Дополнительные факты
Синдром маски Кабуки (синдром макияжа Кабуки) получил свое название от внешнего вида и выражения лица пациентов, придавая им сходство с персонажами японского театра с таким же названием. Известно, что актеры театра Кабуки по-особенному наносят макияж на свои лица, в частности, выделяя глаза и брови, и на пике спектакли замирают на месте, глядя в точку и уменьшая глаза на переносице. Патология была описана в 1981 году двумя авторами, которые представили наблюдения за пациентами с характерными фенотипическими признаками, и в их честь также называется синдромом Ниикава-Куроки. Синдром относится к категории детей-сирот: в Японии его частота составляет 1: 32000, на западе - 1: 86000. На сегодняшний день описано около 350 случаев заболевания.
Причины
Патология имеет гетерогенную этиологию. Синдром, как известно, врожденный. Большинство диагностируемых случаев носят спорадический характер и возникают в результате мутаций, возникающих «de novo». Факторы, влияющие на изменчивость, не определены. В 2010-2012 гг. Изучены две наиболее часто регистрируемые наследственные формы синдрома на основе генных мутаций:
Ген кодирует лизин-специфическую метилтрансферазу 2D, которая находится в 12q13.12. Мутация вызывает заболевание первого типа (KS1) с аутосомно-доминантным наследованием. Встречается у 70% пациентов.
Ген расположен в сайте Xp11.3, кодирует лизин-специфическую 6А-деметилазу и действует как кофактор KMT2. Генетический дефект связан с развитием синдрома второго типа (KS2), наследование которого связано с Х-хромосомой. На его долю приходится около 5% случаев патологии.
В генетических исследованиях есть доказательства сочетания синдрома Кабуки с другими наследственными заболеваниями: синдром Фрейзера, периодический паралич гипокалиемии, синдром Фанкони, сахарный диабет 1 типа. Сообщается, что пациенты структурные перестройки хромосом, такие как хромосома X-кольца, транслокация, дупликация и инверсия хромосомы 8. Относительно небольшая выборка пациентов и относительно недавнее внедрение методов молекулярной цитогенетической диагностики открывают перспективы для дальнейших исследований этиологии синдрома.
Ген кодирует лизин-специфическую метилтрансферазу 2D, которая находится в 12q13.12. Мутация вызывает заболевание первого типа (KS1) с аутосомно-доминантным наследованием. Встречается у 70% пациентов.
Ген расположен в сайте Xp11.3, кодирует лизин-специфическую 6А-деметилазу и действует как кофактор KMT2. Генетический дефект связан с развитием синдрома второго типа (KS2), наследование которого связано с Х-хромосомой. На его долю приходится около 5% случаев патологии.
В генетических исследованиях есть доказательства сочетания синдрома Кабуки с другими наследственными заболеваниями: синдром Фрейзера, периодический паралич гипокалиемии, синдром Фанкони, сахарный диабет 1 типа. Сообщается, что пациенты структурные перестройки хромосом, такие как хромосома X-кольца, транслокация, дупликация и инверсия хромосомы 8. Относительно небольшая выборка пациентов и относительно недавнее внедрение методов молекулярной цитогенетической диагностики открывают перспективы для дальнейших исследований этиологии синдрома.
Патогенез
Из-за неполного изучения причин синдрома Ниикава-Куроки патологические механизмы также остаются недостаточно ясными. Роль и влияние мутаций в генах KMT2D и KDM6A наиболее просвещены. Оба гена действуют как эпигенетические регуляторы модификации гистонов (ядерный белок) и играют важную роль в экспрессии генов. Дефекты кодируемых ими ферментов изменяют метилирование лизина в гистоне Н3, что приводит к развитию гетерогенных врожденных (черепно-лицевых, скелетных, висцеральных), неврологических и эндокринных аномалий. Кроме того, KMT2D и KDM6A участвуют в дифференцировке наивных сD4 + T-лимфоцитов в иммунокомпетентные клетки. Это объясняет тот факт, что многие пациенты с синдромом Кабуки имеют иммунные расстройства, что проявляется в снижении защиты от инфекций и аутоиммунных расстройств.
Клиническая картина
Клиническая картина характеризуется множественными дефектами, касающихся различных анатомических систем: скелет лица, костно-мышечной системы, кожи, сердечно-сосудистой системы, желудочно-кишечного тракта, мочеполовых органов, анализ оборудования Есть также нарушения Определен неврологический, эндокринный и иммунный статус различной степени тяжести. Наиболее патогномоничен для синдрома лицевого дисморфизма, при котором лица пациентов похожи на маски актеров театра Кабуки, невысокого роста и сниженного интеллекта.
Лицевые аномалии включают микроцефалию, очень приподнятые брови (изогнутые), гипертелоризм, глазную щель антимонголоида (провисание наружных углов глаз изнутри), эктропион нижнего века. Портрет пациента с синдромом Кабуки дополняется низкими выпуклыми ушами, широким уплощенным носом, маленьким ртом с тонкими губами. Реже наблюдаются синеватые изменения цвета склеры, расщелины нёба, расщелины губы, микрогнатия, аномалии зубов (гиодонтия, микродонтия, неправильный прикус). Особое выражение лица уже заметно в раннем детстве, с годами черты приобретают еще более грубый и гротескный вид.
Отклонения в развитии костно-суставной системы представлены деформацией позвоночника, сколиозом, недоразвитием трубчатой кости, полиморфными аномалиями пальцев (брахидактилия, синдактилия, арахнодактилия, клинодактилия) и деформацией стопы. На первом году жизни наблюдается задержка роста, в последующем задержка роста сохраняется и становится более заметной. Характерна гипермобильность суставов, что является причиной частых вывихов. Со стороны кожи наблюдается гиперэластичность кожи, наличие гипо- и гиперпигментированных пятен, гиперемия лица, алопеция и гирсутизм. Важным дерматоглифическим маркером являются выступающие подушечки пальцев.
Из висцеральных пороков развития при синдроме Кабуки наиболее распространенными и клинически значимыми являются ИБС (дефект перегородки, однокамерный стеноз, аортальный стеноз, тетрада Фалло, транспозиция крупных сосудов), желудочно-кишечные аномалии (неполное движение кишечника, анальная непроходимость, врожденная диафрагма мочевыводящих путей), гидронефроз, подковообразная почка). У девочек наблюдается преждевременный теларх, у мальчиков - крипторхизм, гидроцеле и микропенис.
Анатомические дефекты сопровождаются функциональными расстройствами - синдромы мальабсорбции и холестаза, блокада сердца, отток мочеточника. Патология зрительного анализатора включает миопию, астигматизм, катаракту, опущенные веки, косоглазие. У большинства детей была диагностирована потеря слуха.
У 80% пациентов выявляются неврологические нарушения: мышечная гипотензия, тремор, судороги, эпилепсия, ЗПРР. Степень олигофрении варьируется от легкой до умеренной. Можно наблюдать особенности аутистического поведения, обсессивно-компульсивного синдрома.
Из-за нарушений иммунной регуляции и снижения уровня сывороточного Ig отмечаются частые инфекции, аутоиммунные патологии (тромбоцитопеническая пурпура, геморрагический васкулит), гематологические заболевания (гемолитическая анемия). Среди гормональных нарушений наиболее распространен сахарный диабет 1 типа, врожденный гипотиреоз, снижение секреции СТГ и АКТГ, ожирение.
Ассоциированные симптомы: Деформация пальцев рук. Нейтропения. Огрубление черт лица. Судороги. Судороги в ногах. Тремор.
Лицевые аномалии включают микроцефалию, очень приподнятые брови (изогнутые), гипертелоризм, глазную щель антимонголоида (провисание наружных углов глаз изнутри), эктропион нижнего века. Портрет пациента с синдромом Кабуки дополняется низкими выпуклыми ушами, широким уплощенным носом, маленьким ртом с тонкими губами. Реже наблюдаются синеватые изменения цвета склеры, расщелины нёба, расщелины губы, микрогнатия, аномалии зубов (гиодонтия, микродонтия, неправильный прикус). Особое выражение лица уже заметно в раннем детстве, с годами черты приобретают еще более грубый и гротескный вид.
Отклонения в развитии костно-суставной системы представлены деформацией позвоночника, сколиозом, недоразвитием трубчатой кости, полиморфными аномалиями пальцев (брахидактилия, синдактилия, арахнодактилия, клинодактилия) и деформацией стопы. На первом году жизни наблюдается задержка роста, в последующем задержка роста сохраняется и становится более заметной. Характерна гипермобильность суставов, что является причиной частых вывихов. Со стороны кожи наблюдается гиперэластичность кожи, наличие гипо- и гиперпигментированных пятен, гиперемия лица, алопеция и гирсутизм. Важным дерматоглифическим маркером являются выступающие подушечки пальцев.
Из висцеральных пороков развития при синдроме Кабуки наиболее распространенными и клинически значимыми являются ИБС (дефект перегородки, однокамерный стеноз, аортальный стеноз, тетрада Фалло, транспозиция крупных сосудов), желудочно-кишечные аномалии (неполное движение кишечника, анальная непроходимость, врожденная диафрагма мочевыводящих путей), гидронефроз, подковообразная почка). У девочек наблюдается преждевременный теларх, у мальчиков - крипторхизм, гидроцеле и микропенис.
Анатомические дефекты сопровождаются функциональными расстройствами - синдромы мальабсорбции и холестаза, блокада сердца, отток мочеточника. Патология зрительного анализатора включает миопию, астигматизм, катаракту, опущенные веки, косоглазие. У большинства детей была диагностирована потеря слуха.
У 80% пациентов выявляются неврологические нарушения: мышечная гипотензия, тремор, судороги, эпилепсия, ЗПРР. Степень олигофрении варьируется от легкой до умеренной. Можно наблюдать особенности аутистического поведения, обсессивно-компульсивного синдрома.
Из-за нарушений иммунной регуляции и снижения уровня сывороточного Ig отмечаются частые инфекции, аутоиммунные патологии (тромбоцитопеническая пурпура, геморрагический васкулит), гематологические заболевания (гемолитическая анемия). Среди гормональных нарушений наиболее распространен сахарный диабет 1 типа, врожденный гипотиреоз, снижение секреции СТГ и АКТГ, ожирение.
Ассоциированные симптомы: Деформация пальцев рук. Нейтропения. Огрубление черт лица. Судороги. Судороги в ногах. Тремор.
Возможные осложнения
Возможные осложнения тесно связаны с многосистемными поражениями ребенка. У новорожденных возможны серьезные нарушения всасывания и глотания, которые требуют установки желудочной трубки или гастростомии. Необъяснимые эпизоды гипогликемии, которые угрожают переходом к гипогликемической коме, также обнаруживаются в младенчестве. Врожденные пороки сердца могут вызвать серьезные нарушения гемодинамики, которые требуют операции на сердце. Распространенные острые респираторные вирусные инфекции часто осложняются отитом или пневмонией. Прогрессирующая атрофия зрительных нервов может привести к слепоте. Пациенты с синдромом Кабуки имеют повышенный риск развития лимфобластного лейкоза и неходжкинской лимфомы.
|
|
Диагностика
Основной диагностической проблемой является низкая осведомленность специалистов первичной медицинской помощи об этом синдроме, отсутствие четко разработанных клинических критериев и методов лабораторной проверки заболевания. Диагноз в основном ставится на основе фенотипических маркеров; крупные медицинские центры предлагают генетическое тестирование. Первоначальный диагноз проводится генетиком, однако пациенты с синдромом Кабуки требуют осмотра и контроля со стороны ряда специалистов: педиатра, кардиолога, невролога, ортопеда, уролога, иммунолога, ортодонта Этапы обследования включают в себя:
• Портретная диагностика. Стигма лица, замедление роста и скелетные аномалии составляют ядро признаков. Помимо высокочастотных проявлений, учитываются также редкие атипичные признаки, известные из описаний отдельных клинических наблюдений (витилиго, множественные гемангиомы, гипоспадия).
• Генетические исследования. Проводится в специализированных лабораторных центрах. Изучите мутационный статус генов MLL2 и KDM6A. Методы флуоресценции (FISH) и сравнительной геномной гибридизации на микрочипах (aCGH) используются для проведения высокоточной диагностики. Проверьте родителей на подобные мутации.
• Лабораторная диагностика. Гемограмма характеризуется тромбоцитопенией, лейкопенией, нейтропенией. Гормональные и иммунологические тесты проводятся для выявления сопутствующих заболеваний. Гликемический профиль, уровень гормонов щитовидной железы и гипофиза, иммуноглобулины, ЦИК.
• Инструментальное исследование. Это делается для выявления сопутствующих заболеваний органов. Первичный диагноз включает эхокардиографию, УЗИ брюшной полости и почек, рентгенографию костей и позвоночника. Видео ЭЭГ-мониторинг проводится с эписиндромом. Кроме того, офтальмологическое обследование, аудиометрия.
При высоком риске рождения ребенка с генетическими отклонениями назначается инвазивный пренатальный диагноз с последующим секвенированием по всему геному. Дифференциальная диагностика проводится с другими генетическими синдромами, сопровождающимися множественными нарушениями развития: Ди Джорджи, Ван дер Вуд, Миллер-Фрейзер, Элерс-Данло, Чардж и другие.
• Портретная диагностика. Стигма лица, замедление роста и скелетные аномалии составляют ядро признаков. Помимо высокочастотных проявлений, учитываются также редкие атипичные признаки, известные из описаний отдельных клинических наблюдений (витилиго, множественные гемангиомы, гипоспадия).
• Генетические исследования. Проводится в специализированных лабораторных центрах. Изучите мутационный статус генов MLL2 и KDM6A. Методы флуоресценции (FISH) и сравнительной геномной гибридизации на микрочипах (aCGH) используются для проведения высокоточной диагностики. Проверьте родителей на подобные мутации.
• Лабораторная диагностика. Гемограмма характеризуется тромбоцитопенией, лейкопенией, нейтропенией. Гормональные и иммунологические тесты проводятся для выявления сопутствующих заболеваний. Гликемический профиль, уровень гормонов щитовидной железы и гипофиза, иммуноглобулины, ЦИК.
• Инструментальное исследование. Это делается для выявления сопутствующих заболеваний органов. Первичный диагноз включает эхокардиографию, УЗИ брюшной полости и почек, рентгенографию костей и позвоночника. Видео ЭЭГ-мониторинг проводится с эписиндромом. Кроме того, офтальмологическое обследование, аудиометрия.
При высоком риске рождения ребенка с генетическими отклонениями назначается инвазивный пренатальный диагноз с последующим секвенированием по всему геному. Дифференциальная диагностика проводится с другими генетическими синдромами, сопровождающимися множественными нарушениями развития: Ди Джорджи, Ван дер Вуд, Миллер-Фрейзер, Элерс-Данло, Чардж и другие.
Лечение
Из-за генетического детерминизма заболевания полное излечение невозможно. Стандарты лечения синдрома не регламентированы; на практике применяется симптоматический подход для устранения тех расстройств, которые мешают развитию ребенка. Хирургическая патология (грыжи, ИБС, расщелина ХЛО, анальная атрезия, крипторхизм ) Устраняются при хирургическом лечении.
Сопутствующие аутоиммунные нарушения корректируются назначением глюкокортикоидов, иммунодепрессантов, моноклональных антител. Рецидивирующие инфекционные процессы диктуют необходимость применения антибактериальных средств, внутривенного введения человеческого иммуноглобулина. При выявлении сахарного диабета 1 типа, вопрос назначения инсулинотерапии решается.
Сопутствующие аутоиммунные нарушения корректируются назначением глюкокортикоидов, иммунодепрессантов, моноклональных антител. Рецидивирующие инфекционные процессы диктуют необходимость применения антибактериальных средств, внутривенного введения человеческого иммуноглобулина. При выявлении сахарного диабета 1 типа, вопрос назначения инсулинотерапии решается.
Список литературы
1. Синдром Кабуки (лекция)/ Боровикова Н.Ю., Мухин Ю., Миронов М.Б., Боровиков С. Русский журнал детской неврологии. 2011.
2. Клинический случай синдрома Кабуки, ассоциированного с эпилепсией/ Боровикова Н.Ю., Боровиков С., Мухин Ю., Миронов М.Б. Эпилепсия и пароксизмальные состояния. 2011.
3. Синдром Кабуки: случай из практики/ Коталевская Ю.Ю., Демикова Н.С. Педиатрия. Журнал им. Г.Н. Сперанского. 2010.
2. Клинический случай синдрома Кабуки, ассоциированного с эпилепсией/ Боровикова Н.Ю., Боровиков С., Мухин Ю., Миронов М.Б. Эпилепсия и пароксизмальные состояния. 2011.
3. Синдром Кабуки: случай из практики/ Коталевская Ю.Ю., Демикова Н.С. Педиатрия. Журнал им. Г.Н. Сперанского. 2010.