Другие названия и синонимы
Ray 's syndrome.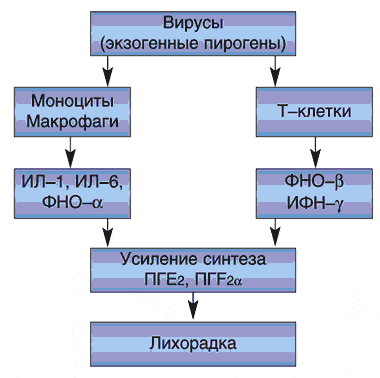
МКБ-10 коды
- МКБ-10
- G93.7 Синдром Рейе
|
|
Описание
Синдром Рея. Быстро прогрессирующая, жизненно угрожающая острая энцефалопатия, сочетающаяся с поражением печени и в классическом варианте обусловленная приемом ацетилсалициловой кислоты на фоне вирусного инфекционного заболевания. Манифестирует внезапной рвотой, затем возникает психомоторное возбуждение, сменяющееся апатией, заторможенностью, дезориентацией с переходом в кому. Диагностируется по клиническим данным с учетом анамнеза, результатов биохимического и клинического анализов крови, исследования ликвора, УЗИ брюшной полости, коагулограммы, пункционной биопсии печени. Основу лечения составляет интенсивная терапия гемодинамических, коагуляционных, дыхательных нарушений и отека мозга. Чем ранее начато лечение, тем больше надежд на благоприятный исход.
Дополнительные факты
Синдром Рея (белая печеночная болезнь, синдром Рейе) - острая быстро прогрессирующая энцефалопатия, возникающая у детей до 16-летнего возраста. Единичные случаи описаны у взрослых. Впервые полное изложение клиники синдрома дал в 1963 г. австралийский патологоанатом Reye, после чего заболевание стало носить его имя и было признано самостоятельной нозологией. Первоначально специалистами в области неврологии и педиатрии синдром Рея ассоциировался только со случаями острой энцефалопатии, возникающей после приема аспирина на фоне вирусной инфекции. Позже были описаны т. н. Рея-подобные заболевания. В связи с этим в настоящее время классифицируют аспирин-ассоциированный, или классический синдром Рея и атипичный синдром Рея, включающий Рея-подобные синдромы.
Наиболее часто синдром встречается среди детей в возрасте от 5 до 14 лет, не зависимо от пола ребенка. Заболеваемость варьирует: в Великобритании у детей до 18 лет она составляет 0,1 случай на 100 тыс. , в США - 1 случай на 100 тыс. В 80-е годы отмечалось существенное снижение заболеваемости после введения ограничений на применение аспирина у детей.
Наиболее часто синдром встречается среди детей в возрасте от 5 до 14 лет, не зависимо от пола ребенка. Заболеваемость варьирует: в Великобритании у детей до 18 лет она составляет 0,1 случай на 100 тыс. , в США - 1 случай на 100 тыс. В 80-е годы отмечалось существенное снижение заболеваемости после введения ограничений на применение аспирина у детей.
Причины
Этиопатогенез синдрома до конца не исследован. Классический синдром Рея развивается в результате приема в качестве антипиретиков препаратов салициловой кислоты детьми, заболевшими ОРВИ и другими вирусными инфекциями (чаще гриппом, ветряной оспой, герпесом, парагриппом). Следует подчеркнуть, что речь идет не о передозировке салицилатов, а об их приеме в пределах рекомендованных возрастных дозировок. Этиофакторами Рея-подобных синдромов могут выступать другие медикаменты, гипоглицин, вальпроат, эндотоксины бактерий, инсектициды и прочие вещества, действующие как митохондриальные яды. По данным проведенных в Великобритании исследований, в 10% случаев синдром Рея сопровождался врожденными метаболическими нарушениями (дисметаболизмом жирных кислот, дефицитом карнитинтрансферазы, нарушениями в цикле мочевины, недостатком глицеринкиназы и тд ).
Главным патогенетическим субстратом синдрома Рея является расстройство функционирования митохондрий, приводящее к клеточному апоптозу (гибели). Триггерами митохондриальных нарушений могут выступать различные экзогенные (медикаменты и тд вещества) и эндогенные (дисметаболические расстройства) факторы. Высказано предположение о том, что синдром Рея возникает у детей, имеющих врожденную субклиническую недостаточность митохондриальных ферментов. Известно, что после всасывания салицилатов и тд веществ их метаболиты попадают в печень, где оказывают токсическое воздействие на митохондрии. Результатом является апоптоз гепатоцитов, жировая инфильтрация печени и нарушение ее детоксикационной функции. Это сопровождается резким повышением концентрации печеночных трансаминаз и аммиака в крови, в несколько раз превышающей норму. Указанные метаболиты оказывают токсическое действие на церебральные ткани, приводя к развитию быстро прогрессирующей токсической энцефалопатии и отеку головного мозга.
Морфологически синдром Рея характеризуется мелко-капельным жировым гепатозом, имеющим распространенный характер и особенно проявленным по периферии печеночных долек. Выраженная в более легкой степени жировая дистрофия наблюдается в других соматических органах: поджелудочной железе, сердечной мышце, эпителии почечных канальцев. В церебральных тканях отмечаются дегенеративные изменения нейронов и отек астроцитов.
Главным патогенетическим субстратом синдрома Рея является расстройство функционирования митохондрий, приводящее к клеточному апоптозу (гибели). Триггерами митохондриальных нарушений могут выступать различные экзогенные (медикаменты и тд вещества) и эндогенные (дисметаболические расстройства) факторы. Высказано предположение о том, что синдром Рея возникает у детей, имеющих врожденную субклиническую недостаточность митохондриальных ферментов. Известно, что после всасывания салицилатов и тд веществ их метаболиты попадают в печень, где оказывают токсическое воздействие на митохондрии. Результатом является апоптоз гепатоцитов, жировая инфильтрация печени и нарушение ее детоксикационной функции. Это сопровождается резким повышением концентрации печеночных трансаминаз и аммиака в крови, в несколько раз превышающей норму. Указанные метаболиты оказывают токсическое действие на церебральные ткани, приводя к развитию быстро прогрессирующей токсической энцефалопатии и отеку головного мозга.
Морфологически синдром Рея характеризуется мелко-капельным жировым гепатозом, имеющим распространенный характер и особенно проявленным по периферии печеночных долек. Выраженная в более легкой степени жировая дистрофия наблюдается в других соматических органах: поджелудочной железе, сердечной мышце, эпителии почечных канальцев. В церебральных тканях отмечаются дегенеративные изменения нейронов и отек астроцитов.
Клиническая картина
Классический синдром Рея в среднем манифестирует через 3 суток после перенесенной вирусной инфекции, хотя этот период может колебаться от 12 ч до 21 дня. При ветряной оспе дебют синдрома приходится на 4-5 день сыпи. Обязательным симптомом является рвота. Как правило, она носит многократный характер. Через 24-48 ч после начала рвоты отмечаются изменения в поведении ребенка, варьирующие от раздражительности и необычного возбуждения до заторможенности и сонливости. Ребенок перестает разговаривать, не хочет есть и пить, контакт с ним затруднен. Достаточно быстро больной перестает ориентироваться в окружающей обстановке и во времени, возникает делирий. Эти состояния сопровождаются гипервентиляцией. Затем ребенок впадает в кому, которая вначале имеет интермиттирующий тип и продолжается не более 3 а потом может длиться от 1-4 суток до нескольких недель. В последней стадии синдрома возникает остановка дыхания.
Диагностика
Заподозрить синдром Рея педиатру или неврологу позволяют выявление в анамнезе связи с вирусной инфекцией, приемом аспирина и других источников митохондриальных токсинов, типичная клиника заболевания (рвота с последующими психо-неврологическими нарушениями), увеличение печени. Верифицировать синдром Рея помогает биохимический анализ крови с определением уровня печеночных ферментов, УЗИ брюшной полости, биопсия печени, исследование ликвора.
Биохимия крови констатирует увеличение АСТ и АЛТ в 3 и более раз (в ряде случаев в 20 раз) при нормальном содержании билирубина. Патогномоничным признаком является резкое повышение уровня аммиака. У 40% заболевших наблюдается гипогликемия, как правило, это дети до 5-летнего возраста. Концентрация электролитов крови может изменяться соответственно степени дегидратации, развивающейся вследствие рвоты и отказа от питья. В большинстве случаев синдром Рея сопровождается нарушением свертывающей системы, что находит отражение в данных коагулограммы. Клинический анализ крови остается в пределах нормы, иногда наблюдается некоторое увеличение лейкоцитов.
С целью исключения инфекционной патологии ЦНС проводится люмбальная пункция. Исследование цереброспинальной жидкости свидетельствует о ее стерильности и не выявляет существенных отклонений в ее составе, что позволяет исключить инфекционно-воспалительный характер энцефалопатии. УЗИ органов брюшной полости определяет увеличение печени - гепатомегалию, ее диффузно повышенную эхогенность и структурную уплотненность; возможны подобные изменения поджелудочной железы. Если синдром Рея не может быть установлен по данным вышеперечисленных методов диагностики, проводится биопсия печени. Морфологическое исследование биоптатов выявляет типичную для синдрома картину: отсутствие воспалительных изменений и наличие признаков выраженной жировой дистрофии.
Синдром Рея является диагнозом исключения.
Биохимия крови констатирует увеличение АСТ и АЛТ в 3 и более раз (в ряде случаев в 20 раз) при нормальном содержании билирубина. Патогномоничным признаком является резкое повышение уровня аммиака. У 40% заболевших наблюдается гипогликемия, как правило, это дети до 5-летнего возраста. Концентрация электролитов крови может изменяться соответственно степени дегидратации, развивающейся вследствие рвоты и отказа от питья. В большинстве случаев синдром Рея сопровождается нарушением свертывающей системы, что находит отражение в данных коагулограммы. Клинический анализ крови остается в пределах нормы, иногда наблюдается некоторое увеличение лейкоцитов.
С целью исключения инфекционной патологии ЦНС проводится люмбальная пункция. Исследование цереброспинальной жидкости свидетельствует о ее стерильности и не выявляет существенных отклонений в ее составе, что позволяет исключить инфекционно-воспалительный характер энцефалопатии. УЗИ органов брюшной полости определяет увеличение печени - гепатомегалию, ее диффузно повышенную эхогенность и структурную уплотненность; возможны подобные изменения поджелудочной железы. Если синдром Рея не может быть установлен по данным вышеперечисленных методов диагностики, проводится биопсия печени. Морфологическое исследование биоптатов выявляет типичную для синдрома картину: отсутствие воспалительных изменений и наличие признаков выраженной жировой дистрофии.
Синдром Рея является диагнозом исключения.
Дифференциальная диагностика
Дифференциальная диагностика проводится с вирусными инфекциями, энцефалитом, менингитом, различными интоксикационными синдромами, субарахноидальным кровоизлиянием и пр. После выздоровления дети, перенесшие синдром, направляются на консультацию генетика для исключения наличия у них врожденных метаболических заболеваний.
|
|
Лечение
Подозрение на синдром Рея является показанием к срочной госпитализации в отделение интенсивной терапии. Лечение является скорее симптоматическим и направлено на купирование происходящих в организме патологических процессов и поддержание жизненно важных органов. Применяют кортикостероиды (в основном преднизолон), инфузии электролитов и витамина маннитол для купирования отека мозга. Внутривенное введение жидкости с одной стороны необходимо для скорейшего выведения аммиака и прочих токсических веществ из организма, а с другой стороны ограниченно из-за опасности прогрессирования церебрального отека. Осуществляется коррекция нарушений кровообращения и расстройств гемостаза. При дыхательных нарушениях производят интубацию трахеи с гипервентиляцией. Лечение осуществляется при постоянном мониторинге артериального давления, газового состава крови, внутричерепного давления.
Смертность на начальной стадии синдрома составляет 5%, в стадии интермиттирующей комы - 50-60% , на последней стадии - 95%. Причиной летального исхода обычно выступает отек мозга, реже - дыхательная недостаточность, желудочное кровотечение вследствие коагулопатии, сердечная недостаточность, сепсис или ОПН. Если синдром Рея распознан в ранней стадии и незамедлительно начата соответствующая терапия, то можно надеяться на благополучный исход. Благодаря совершенствованию методов диагностики и лечения показатель летальности удалось снизить с 40%, регистрируемых в 1970-х гг. до 20% в 1990-х гг. У выживших детей, как правило, отмечается полное нейро-психическое восстановление.
Смертность на начальной стадии синдрома составляет 5%, в стадии интермиттирующей комы - 50-60% , на последней стадии - 95%. Причиной летального исхода обычно выступает отек мозга, реже - дыхательная недостаточность, желудочное кровотечение вследствие коагулопатии, сердечная недостаточность, сепсис или ОПН. Если синдром Рея распознан в ранней стадии и незамедлительно начата соответствующая терапия, то можно надеяться на благополучный исход. Благодаря совершенствованию методов диагностики и лечения показатель летальности удалось снизить с 40%, регистрируемых в 1970-х гг. до 20% в 1990-х гг. У выживших детей, как правило, отмечается полное нейро-психическое восстановление.
Прогноз
Предупредить возникновение синдрома позволяет отказ от применения салицилатов у детей. В настоящее время во многих странах проводится политика замены салицилатов препаратами парацетамола или ибупрофена при необходимости проведения антипиретической терапии.