ICD-10 codes
- ICD-10
- G71.2 Congenital myopathies
Description
Врожденная парамиотония. Отдельный клинический вариант миотонии, наследуемый аутосомно - доминантным путем и характеризующийся возникновением миотонических эпизодов под воздействием холода. В клинике заболевания также присутствуют пароксизмы транзиторной мышечной слабости. Диагностический алгоритм включает оценку неврологического статуса, биохимическое исследование крови, электронейромиографию, генеалогический анализ, при необходимости - поиск мутации в гене SCN4A. Лечение симптоматическое. Возможно применение местных анестетиков и диуретиков. Прогноз для жизни благоприятный.
Additional facts
Врожденная парамиотония - вариант миотонии, при котором основным провоцирующим триггером миотонических феноменов выступает холод. Описана А. Эйленбургом в 1886 г. , в связи с чем носит название парамиотония Эйленбурга. М. Левандовский в 1916 г. дал описание врожденной парамиотонии под названием «холодовый паралич». В литературе по неврологии можно также встретить термины «холодовая миотония» и «парадоксальная миотония». Последнее название связано с типичным для парамиотонии усилением миотонических проявлений на фоне двигательной активности, в отличии от классической миотонии Томсена, при которой повторные движения приводят к уменьшению ригидности и нормализации работы мышц.
Врожденная парамиотония встречается с частотой 1 случай на 100 тыс. человек. Является генетически обусловленной каналопатией, передается по наследству аутосомно-доминантно. Дебютирует на первом десятилетии жизни, в большинстве случаев в первый год от рождения ребенка. Заболеваемость не имеет гендерных предпочтений.
Врожденная парамиотония встречается с частотой 1 случай на 100 тыс. человек. Является генетически обусловленной каналопатией, передается по наследству аутосомно-доминантно. Дебютирует на первом десятилетии жизни, в большинстве случаев в первый год от рождения ребенка. Заболеваемость не имеет гендерных предпочтений.
Reasons
Этиопатогенетическим субстратом парамиотонии является дефект гена SCN4A (17-я хромосома, локус 17q23. 1-q25. 3), отвечающего за кодирование α-субъединиц натриевых каналов скелетной мускулатуры. Генетические аберрации в данном локусе обусловливают также возникновение пароксизмальной миоплегии. В случае парамиотонии дефект в гене вызывает повышение уровня и увеличение длительности деполяризации плазматической мембраны мышечного волокна (миофибриллы). Следствием этого является затруднение расслабления мышцы (мышечная ригидность) из-за длительно не спадающего напряжения составляющих ее миофибрилл. При слишком высоком уровне деполяризации мышца утрачивает свою способность к возбудимости, что клинически проявляется в виде пареза - мышечной слабости. Провоцирующими триггерами миотонического пароксизма выступают общее и локальное охлаждение, а также избыточное поступление калия в организм.
Symptoms
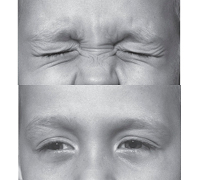
Заболевание может проявляться начиная с периода новорожденности. Так, опытные матери отмечают слишком долгое закрывание глаз у ребенка при умывании его холодной водой. Миотонический феномен состоит в удлиненной фазе тонического сокращения мышцы, ее замедленном расслаблении. При врожденной парамиотонии он провоцируется прохладной погодой, понижением температуры окружающего воздуха до 10-12 градусов, местным охлаждением (мытье рук холодной водой, прикладывание льда, употребление в пищу мороженного ), присутствием в рационе богатых калием продуктов.
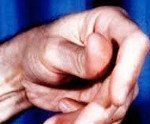
Миотонические спазмы наблюдаются в отдельных мышечных группах. Наиболее часто страдает лицевая мускулатура - мышца века, круговая мышца рта, жевательные мышцы. Миотонии могут локализоваться в мышцах шеи, рук, ног, языка, глотки. Длительность миотонического сокращения мышц варьирует от 15 мин до нескольких часов. Патогномоничным признаком выступает усиление миотонического феномена на фоне повторных движений с участием пораженных мышц и уменьшение миотонии при согревании. В ряде случаев холод, наряду с миотонией, провоцирует развитие мышечной слабости (преходящего пареза), которая может длиться до нескольких суток.
Миотонические спазмы наблюдаются в отдельных мышечных группах. Наиболее часто страдает лицевая мускулатура - мышца века, круговая мышца рта, жевательные мышцы. Миотонии могут локализоваться в мышцах шеи, рук, ног, языка, глотки. Длительность миотонического сокращения мышц варьирует от 15 мин до нескольких часов. Патогномоничным признаком выступает усиление миотонического феномена на фоне повторных движений с участием пораженных мышц и уменьшение миотонии при согревании. В ряде случаев холод, наряду с миотонией, провоцирует развитие мышечной слабости (преходящего пареза), которая может длиться до нескольких суток.
Diagnostics
Начало в раннем детском возрасте и типичная клиника позволяют педиатру заподозрить врожденную нервно-мышечную патологию и направить ребенка к неврологу. При постановке диагноза последний опирается на клинические данные, результаты неврологического осмотра, ЭФИ нервно-мышечной системы, биохимического анализа крови, генетических исследований.
Электронейромиография или стимуляционная электромиография позволяют определить сохранность проведения нервных импульсов по нервным стволам и установить первично-мышечный тип поражения нервно-мышечного аппарата. В анализе крови определяется нормальный уровень креатинфосфокиназы (КФК), что дает возможность дифференцировать парамиотонию от прогрессирующих мышечных дистрофий. Исследование генеалогического древа, проводимое генетиком, помогает проследить аутосомно-доминантный путь наследования. При помощи ДНК-анализа возможно выявление мутации в гене SCN4A, однако это достаточно дорогостоящее исследование.
Электронейромиография или стимуляционная электромиография позволяют определить сохранность проведения нервных импульсов по нервным стволам и установить первично-мышечный тип поражения нервно-мышечного аппарата. В анализе крови определяется нормальный уровень креатинфосфокиназы (КФК), что дает возможность дифференцировать парамиотонию от прогрессирующих мышечных дистрофий. Исследование генеалогического древа, проводимое генетиком, помогает проследить аутосомно-доминантный путь наследования. При помощи ДНК-анализа возможно выявление мутации в гене SCN4A, однако это достаточно дорогостоящее исследование.
Treatment
Эффективная терапия пока не найдена. Лечение имеет симптоматический характер. По мнению ряда авторов результативно применение местных анестетиков, обладающих способностью угнетать трансмембранный ток натрия. Препаратом выбора выступает мексилетин в капсулах, который, в отличие от лидокаина, хорошо усваивается при приеме внутрь. Исследования показали его эффективность в отношении ригидности и мышечной слабости. Для уменьшения частоты и выраженности миотонических пароксизмов могут использоваться диуретические фармпрепараты (ацетазоламид, гидрохлортиазид), применение которых приводит к снижению концентрации калия в крови.
Forecast
Врожденная парамиотония, как правило, характеризуется доброкачественным стабильным течением без прогрессирования. Прогноз для выздоровления неблагоприятный. Однако, не смотря на сохраняющиеся в течение жизни парамиотонические симптомы, продолжительность жизни пациентов не страдает. Больные не теряют способности к труду и самообслуживанию, не имеют проблем с социальной адаптацией.