ICD-10 codes
Description
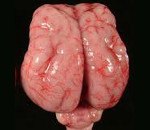
Лиссэнцефалия. Группа генетически обусловленных аномалий развития головного мозга, характеризующихся частичным или полным недоразвитием извилин и борозд коры больших полушарий, а также нарушением ее ультраструктуры. Выраженность и сочетания симптомов этого состояния различаются при разных формах заболевания, наиболее распространены судороги, глубокая умственная отсталость, нарушения глотания и гипотония мышц. Диагностика лиссэнцефалии может производиться ультразвуковыми методиками (в том числе и пренатально), компьютерной и магнитно-резонансной томографией, для наиболее распространенных форм возможно определение посредством молекулярно-генетического анализа. Специфического лечения не существует, используют симптоматическую и поддерживающую терапию.
Additional facts
Лиссэнцефалия - группа тяжелых аномалий развития головного мозга, которые сопровождаются недоразвитием коры больших полушарий с формированием пахигирии (наличием всего нескольких извилин и борозд) или агирии (полным отсутствием складчатости коры). Данная патология может выступать в качестве самостоятельного генетического заболевания или входить в симптомокомплекс других синдромов - например, Миллера-Дикера, Фукуямы и Уокера-Варбурга. Механизм наследования различных типов лиссэнцефалии может быть аутосомно-рецессивным, аутосомно-доминантным (в данном случае чаще всего имеют место спонтанные или герминативные мутации) и сцепленным с Х-хромосомой. Из-за многообразия механизмов наследования половое распределение нарушения неодинаково при различных формах патологии. Лиссэнцефалия является достаточно редкой генетической аномалией развития мозга, поэтому встречаемость определена только для наиболее распространенной первой группы нарушений - она составляет 11,7 случаев на 1 000 000 новорожденных. Для остальных групп лиссэнцефалии встречаемость не установлена, в том числе и потому, что во многих случаях плод с подобной патологией не вынашивается, и беременность самопроизвольно прерывается в первом триместре еще до определения наличия порока.
Symptoms
Ассоциированные симптомы: Судороги. Судороги в ногах.
Reasons
Основная общая причина всех типов лиссэнцефалии - нарушение процесса миграции клеток-предшественников нейронов (нейробластов) из передних отделов нервной трубки к будущей коре больших полушарий. В результате этого вместо сложной складчатой структуры, которая имеет в своем составе шесть слоев, образуется гладкая или имеющая в разы меньше борозд кора, состоящая из 2-4 слоев (в зависимости от формы заболевания). Поскольку кора больших полушарий у человека отвечает за когнитивные функции, в ней содержится огромное число нервных центров и обширные ассоциативные зоны, лиссэнцефалия приводит к тяжелейшим расстройствам. Кроме того, при некоторых типах мутаций, вызывающих данное состояние, возможно развитие аномалий других органов и тканей, что еще больше усугубляет состояние больного.
Наиболее распространенные типы лиссэнцефалии обусловлены дефектами гена PAFAH1B1, также известного под названием LIS1 и расположенного на 17-й хромосоме. Многие врачи-генетики отмечают, что для возникновения выраженного недоразвития коры головного мозга и синдрома Миллера-Дикера необходимы не точечные мутации в LIS1, а крупные делеции с захватом сотен пар азотистых оснований. Нередко при этом повреждаются и окружающие гены, что становится причиной разнообразных фенотипических проявлений данного типа лиссэнцефалии. Ген LIS1 кодирует внутриклеточную субъединицу сложного по своей структуре фермента, который принимает активное участие в миграции нейробластов. Дефекты в структуре этой субъединицы приводят к нарушению данного процесса и развитию данного заболевания.
Другая форма лиссэнцефалии обусловлена мутацией гена DCX, локализованного на Х-хромосоме, поэтому наследование этого типа патологии сцеплено с полом. Белок, получаемый в результате экспрессии данного гена, участвует в формировании особого типа микротрубочек, которые вырабатываются только нейробластами и необходимы им для формирования связей между клетками. Нарушения в структуре DCX приводят к синтезу дефектного белка, что провоцирует лиссэнцефалию. Другой относительно изученный вариант этого состояния, также сцепленный с Х-хромосомой, вызывается мутациями гена ARX, который является фактором транскрипции для иных генов. Он контролирует развитие коры больших полушарий, поджелудочной железы и половых органов, из-за чего дефекты ARX проявляются многочисленными пороками данных органов, в том числе лиссэнцефалией.
Еще одним распространенным вариантом лиссэнцефалии является форма заболевания, обусловленная мутацией гена RELN, расположенного на 7-й хромосоме. Дефекты этого гена приводят к так называемому синдрому Норман-Робертса, который, помимо прочего, сопровождается выраженным нарушением складчатости коры больших полушарий. Ген RELN кодирует последовательность гликопротеида рилина, который принимает активное участие в формировании нервной ткани, а также контролирует образование новых дендритов и функционирование долговременной памяти у взрослых людей. Удалось идентифицировать еще один ген, приводящий к развитию лиссэнцефалии - TUBA1A, который локализован на 12-й хромосоме. Он кодирует определенный компонент цитоскелета нейронов, его дефект приводит к неполноценности нейробластов, что нарушает процесс их миграции в эмбриональном периоде.
Наиболее распространенные типы лиссэнцефалии обусловлены дефектами гена PAFAH1B1, также известного под названием LIS1 и расположенного на 17-й хромосоме. Многие врачи-генетики отмечают, что для возникновения выраженного недоразвития коры головного мозга и синдрома Миллера-Дикера необходимы не точечные мутации в LIS1, а крупные делеции с захватом сотен пар азотистых оснований. Нередко при этом повреждаются и окружающие гены, что становится причиной разнообразных фенотипических проявлений данного типа лиссэнцефалии. Ген LIS1 кодирует внутриклеточную субъединицу сложного по своей структуре фермента, который принимает активное участие в миграции нейробластов. Дефекты в структуре этой субъединицы приводят к нарушению данного процесса и развитию данного заболевания.
Другая форма лиссэнцефалии обусловлена мутацией гена DCX, локализованного на Х-хромосоме, поэтому наследование этого типа патологии сцеплено с полом. Белок, получаемый в результате экспрессии данного гена, участвует в формировании особого типа микротрубочек, которые вырабатываются только нейробластами и необходимы им для формирования связей между клетками. Нарушения в структуре DCX приводят к синтезу дефектного белка, что провоцирует лиссэнцефалию. Другой относительно изученный вариант этого состояния, также сцепленный с Х-хромосомой, вызывается мутациями гена ARX, который является фактором транскрипции для иных генов. Он контролирует развитие коры больших полушарий, поджелудочной железы и половых органов, из-за чего дефекты ARX проявляются многочисленными пороками данных органов, в том числе лиссэнцефалией.
Еще одним распространенным вариантом лиссэнцефалии является форма заболевания, обусловленная мутацией гена RELN, расположенного на 7-й хромосоме. Дефекты этого гена приводят к так называемому синдрому Норман-Робертса, который, помимо прочего, сопровождается выраженным нарушением складчатости коры больших полушарий. Ген RELN кодирует последовательность гликопротеида рилина, который принимает активное участие в формировании нервной ткани, а также контролирует образование новых дендритов и функционирование долговременной памяти у взрослых людей. Удалось идентифицировать еще один ген, приводящий к развитию лиссэнцефалии - TUBA1A, который локализован на 12-й хромосоме. Он кодирует определенный компонент цитоскелета нейронов, его дефект приводит к неполноценности нейробластов, что нарушает процесс их миграции в эмбриональном периоде.
Classification
На сегодняшний день выявлено более двадцати вариантов лиссэнцефалии. Эти варианты обусловлены различными мутациями, имеют отличия в фенотипических проявлениях заболевания, разные механизмы наследования и тяжесть симптомов. Долгое время разновидности патологии не удавалось успешно классифицировать из-за значительного разнообразия генетических дефектов и недостаточных данных о ключевых генах, приводящих к развитию некоторых форм заболевания. Лишь в 2003-м году удалось создать достаточно приемлемую с точки зрения современной генетики и неврологии классификацию лиссэнцефалии, которая учитывает основные нюансы этого состояния. Специалисты разделяют все формы лиссэнцефалии на пять классов:
• Класс 1, часто называемый классической лиссэнцефалией. Включает в себя формы заболевания, обусловленные мутациями гена LIS1 (изолированный тип и синдром Миллера-Дикера), а также сцепленную с полом разновидность, вызванную мутацией DCX. Кроме того, в этот класс часто включают некоторые типы лиссэнцефалии с неясными генетическими причинами. Особенностью группы является нарушение складчатости коры больших полушарий с минимальным проявлением иных аномалий центральной нервной системы.
• Класс 2. В настоящее время состоит только из одного типа лиссэнцефалии, которая вызывается дефектами гена ARX. Заболевание сцеплено с Х-хромосомой, помимо пороков развития коры у больных часто обнаруживаются отсутствие мозолистого тела, нарушения терморегуляции, выраженные аномалии половых органов и поджелудочной железы.
• Класс 3. Включает в себя разновидности лиссэнцефалии в сочетании с гипоплазией или полным недоразвитием мозжечка, наиболее типичной мутацией для данного класса являются дефекты гена RELN. Может регистрироваться как изолированный вариант патологии или в составе синдрома Норман-Робертса.
• Класс 4, нередко называется микролиссэнцефалией, поскольку нарушение формирования коры больших полушарий сопровождается выраженной микроцефалией. Некоторые формы этого заболевания были достоверно ассоциированы с мутациями гена TUBA1A.
• Класс 5 или булыжниковые лиссэнцефалии, к которым относят синдромы Фукуямы и Уокера-Варбурга. Последний соотносится с дефектами генов POMT1, POMT2, FKRP и некоторыми другими, все они расположены на 9-й хромосоме.
Данная классификация критикуется некоторыми исследователями по причине наличия большого количества «белых пятен» в виде включения в нее форм лиссэнцефалии с неопределенными ключевыми генами. Однако в настоящее время именно это разделение считается наиболее общепринятым в научном мире. Классификация корректируется по мере дальнейшего изучения данного состояния.
• Класс 1, часто называемый классической лиссэнцефалией. Включает в себя формы заболевания, обусловленные мутациями гена LIS1 (изолированный тип и синдром Миллера-Дикера), а также сцепленную с полом разновидность, вызванную мутацией DCX. Кроме того, в этот класс часто включают некоторые типы лиссэнцефалии с неясными генетическими причинами. Особенностью группы является нарушение складчатости коры больших полушарий с минимальным проявлением иных аномалий центральной нервной системы.
• Класс 2. В настоящее время состоит только из одного типа лиссэнцефалии, которая вызывается дефектами гена ARX. Заболевание сцеплено с Х-хромосомой, помимо пороков развития коры у больных часто обнаруживаются отсутствие мозолистого тела, нарушения терморегуляции, выраженные аномалии половых органов и поджелудочной железы.
• Класс 3. Включает в себя разновидности лиссэнцефалии в сочетании с гипоплазией или полным недоразвитием мозжечка, наиболее типичной мутацией для данного класса являются дефекты гена RELN. Может регистрироваться как изолированный вариант патологии или в составе синдрома Норман-Робертса.
• Класс 4, нередко называется микролиссэнцефалией, поскольку нарушение формирования коры больших полушарий сопровождается выраженной микроцефалией. Некоторые формы этого заболевания были достоверно ассоциированы с мутациями гена TUBA1A.
• Класс 5 или булыжниковые лиссэнцефалии, к которым относят синдромы Фукуямы и Уокера-Варбурга. Последний соотносится с дефектами генов POMT1, POMT2, FKRP и некоторыми другими, все они расположены на 9-й хромосоме.
Данная классификация критикуется некоторыми исследователями по причине наличия большого количества «белых пятен» в виде включения в нее форм лиссэнцефалии с неопределенными ключевыми генами. Однако в настоящее время именно это разделение считается наиболее общепринятым в научном мире. Классификация корректируется по мере дальнейшего изучения данного состояния.
Diagnostics
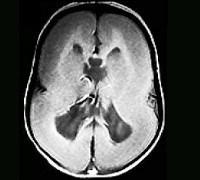
В отношении лиссэнцефалии возможна пренатальная диагностика при помощи ультразвуковых методов исследования, при этом увеличение разрешающей способности УЗИ-аппаратуры способствует все более раннему определению заболевания. Развитие нарушений в строении коры больших полушарий происходит на 14-20-й неделе гестации, в настоящее время уже в этот период можно определить патологию и поставить вопрос о прерывании беременности. После рождения ребенка с подозрением на лиссэнцефалию диагноз подтверждают при помощи КТ и МРТ. Молекулярно-генетическая диагностика обладает высокой точностью, но она доступна только в отношении тех форм заболевания, для которых известны ключевые гены. Специфического лечения лиссэнцефалии не существует, применяют противосудорожные препараты, ноотропы и другие средства, позволяющие уменьшить выраженность симптоматики. При наличии иных пороков развития производится их коррекция по медицинским показаниям.
Treatment
Прогноз практически любой формы лиссэнцефалии крайне неблагоприятный, большинство больных умирает в раннем детстве от осложнений и других пороков развития. Описаны отдельные легкие случаи этого состояния с частичным или очаговым недоразвитием коры больших полушарий, но не все специалисты склонны причислять такие типы патологии к лиссэнцефалиям. Профилактика возможна только в рамках пренатальной диагностики. При наличии в роду подобных заболеваний имеет смысл провести генетический анализ на носительство генов аутосомно-рецессивных типов патологии. При обнаружении у плода нарушений формирования мозга, соответствующих лиссэнцефалии, ставится вопрос о прерывании беременности.