ICD-10 codes
Description
Синдром Пфайффера. Генетическое заболевание с аутосомно - доминантным механизмом наследования, характеризующееся нарушением формирования костей черепа и конечностей. Симптомами являются деформации черепа (в результате краниосиностоза), костей пальцев рук и ног. Некоторые формы заболевания характеризуются глухотой, нарушением интеллектуального развития, стоматологическими патологиями. Диагностика синдрома Пфайффера производится на основании данных настоящего статуса больного, рентгенологических исследований, молекулярно-генетических анализов. Специфическое лечение отсутствует, применяют паллиативные мероприятия для предотвращения тяжелых осложнений (в том числе и хирургического характера) и симптоматическую терапию.
Additional facts
Синдром Пфайффера - врожденное генетически обусловленное нарушение формирования черепа и конечностей, которое характеризуется краниосиностозом, деформацией лица, расширением и укорочением пальцев, частыми синдактилиями. Впервые это заболевание было описано в 1964 году немецким педиатром Рудольфом Пфайффером, в дальнейшем было выявлено несколько клинических форм патологии. По данным современной генетики, синдром Пфайффера является аутосомно-доминантным состоянием с полной пенетрантностью, половое распределение больных не имеет особенностей - с одинаковой частотой болеют как мальчики, так и девочки. Встречаемость точно не определена, по некоторым данным она составляет примерно 1:100 000 новорожденных. Каких-либо национальных или расовых особенностей в распределении этого заболевания также не выявлено. В отношении синдрома Пфайффера крайне важна ранняя диагностика, так как от времени начала паллиативного лечения во многом зависит прогноз заболевания и качество дальнейшей жизни больного.
Reasons
Несмотря на то, что синдром Пфайффера является наследственным заболеванием с аутосомно-доминантным механизмом передачи, большинство случаев этой патологии являются следствием спонтанных герминативных мутаций в половых клетках родителей. При этом за развитие состояния отвечают дефекты двух генов, кодируемые ими белки обладают сходными функциями. Первый ген - FGFR1, расположен на 8-й хромосоме и кодирует последовательность белка-рецептора к фактору роста фибробластов-1. Наиболее частым типом генетического дефекта при этом становится миссенс-мутация 252Pro-Arg в экзоне 7а. Другим геном, мутации которого приводят к развитию синдрома Пфайффера, является FGFR2, расположенный на 10-й хромосоме - продуктом его экспрессии также является рецептор к фактору роста фибробластов (РФРФ-2). В этом гене выявлено более 26 различных вариантов мутаций, способных приводить к различным формам синдрома Пфайффера.
По причине того, что вышеуказанные белки обладают очень похожими функциями, патогенез синдрома Пфайффера при дефектах гена FGFR1 и FGFR2 тоже весьма схож. В результате миссенс-мутаций изменяется структура экспрессируемого белка, из-за чего полученный рецептор становится неспособен полноценно связываться с веществом-стимулятором (фактором роста фибробластов) и передавать информацию внутрь клетки. Так как фибробласты активно участвуют в процессах остеогенеза в эмбриональный период и в первые годы жизни ребенка, такое изменение рецепторов ведет к разнообразным костным патологиям.
Доказательством этого является тот факт, что дефекты гена FGFR1, помимо синдрома Пфайффера, приводят к тригоноцефалии 1-го типа и ряду других нарушений, а FGFR2 - к неспецифическому краниосиностозу, синдрому Крузона и иным похожим патологиям. При синдроме Пфайффера происходит преждевременное сращение костей черепа, что становится причиной деформации головы и лица, повышения внутричерепного давления - это, в свою очередь, способно вызывать ряд вторичных нарушений от нейросенсорных расстройств до глубокой умственной отсталости.
По причине того, что вышеуказанные белки обладают очень похожими функциями, патогенез синдрома Пфайффера при дефектах гена FGFR1 и FGFR2 тоже весьма схож. В результате миссенс-мутаций изменяется структура экспрессируемого белка, из-за чего полученный рецептор становится неспособен полноценно связываться с веществом-стимулятором (фактором роста фибробластов) и передавать информацию внутрь клетки. Так как фибробласты активно участвуют в процессах остеогенеза в эмбриональный период и в первые годы жизни ребенка, такое изменение рецепторов ведет к разнообразным костным патологиям.
Доказательством этого является тот факт, что дефекты гена FGFR1, помимо синдрома Пфайффера, приводят к тригоноцефалии 1-го типа и ряду других нарушений, а FGFR2 - к неспецифическому краниосиностозу, синдрому Крузона и иным похожим патологиям. При синдроме Пфайффера происходит преждевременное сращение костей черепа, что становится причиной деформации головы и лица, повышения внутричерепного давления - это, в свою очередь, способно вызывать ряд вторичных нарушений от нейросенсорных расстройств до глубокой умственной отсталости.
Symptoms
На сегодняшний день выделяют как минимум три клинические формы синдрома Пфайффера, которые различаются между собой выраженностью нарушений, течением и прогнозом заболевания. Причина такого различия лежит в молекулярно-генетических механизмах развития патологии - 1-й тип обусловлен единственной мутацией гена FGFR1 (252Pro-Arg) и некоторыми нарушениями структуры FGFR2, тогда как 2-й и 3-й типы вызваны различными вариантами дефектов только гена FGFR2. Более тонкие особенности патогенеза каждой формы заболевания в настоящее время находятся в стадии изучения. Благодаря характерной клинической картине, специалисты могут определить разновидность синдрома Пфайффера, основываясь только на симптомах патологии. Молекулярно-генетический анализ во многих случаях необходим лишь для окончательного подтверждения диагноза.
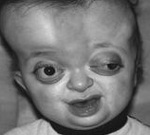
Синдром Пфайффера 1. Го типа является наиболее распространенным и благоприятным в прогностическом отношении вариантом заболевания. При этом состоянии уже с самого рождения ребенка отмечаются аномалии развития лица - гипертелоризм, недоразвитие верхнечелюстных костей, расширенные плоские переносица и спинка носа. Иногда могут отмечаться расщепление твердого нёба и экзофтальм. При этой форме синдрома Пфайффера характерные изменения формы черепа сводятся к увеличению его высоты и длины, очень редко отмечается асимметрия. В дальнейшем могут возникать аномалии зубов - искривление зубного ряда, гипоплазия, склонность к кариесу. Деформации костей конечностей сводятся к расширению фаланг первых пальцев на руках и ногах, может отмечаться частичная синдактилия.
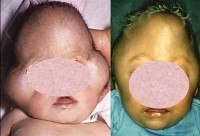
Синдром Пфайффера 2. Го типа - более тяжелая форма заболевания, проявляющаяся значительной выраженностью сращения костей черепа между собой и рядом других аномалий. Голова из-за краниосиностоза приобретает характерную форму «трилистника» или «листка клевера», наблюдается значительная гипоплазия средней трети лица. По причине грубого нарушения формирования черепа данный тип синдрома Пфайффера характеризуется выраженной умственной отсталостью и рядом неврологических нарушений. Пороки развития конечностей сводятся к расширению первых пальцев, синдактилии, анкилозу локтевых суставов. Кроме того, синдром Пфайффера 2-го типа часто сопровождается различными пороками развития внутренних органов. Все вышеперечисленные нарушения могут стать причиной летального исхода в раннем возрасте.
Синдром Пфайффера 3. Го типа во многом схож по своим проявлениям с предыдущим вариантом заболевания, но краниосиностоз при этом не приводит к формированию деформации черепа в виде «листка клевера». У больных отмечается увеличение головы в высоту, пороки развития конечностей (синдактилии, анкилозы суставов), экзофтальм, неврологические нарушения. Для этого варианта синдрома Пфайффера характерно раннее развитие зубов, нередко младенцы уже рождаются с ними (натальные зубы). Также возможны пороки внутренних органов и ранняя смерть из-за совокупности нарушений.
Ассоциированные симптомы: Деформация пальцев ног. Деформация пальцев рук.
Синдром Пфайффера 1. Го типа является наиболее распространенным и благоприятным в прогностическом отношении вариантом заболевания. При этом состоянии уже с самого рождения ребенка отмечаются аномалии развития лица - гипертелоризм, недоразвитие верхнечелюстных костей, расширенные плоские переносица и спинка носа. Иногда могут отмечаться расщепление твердого нёба и экзофтальм. При этой форме синдрома Пфайффера характерные изменения формы черепа сводятся к увеличению его высоты и длины, очень редко отмечается асимметрия. В дальнейшем могут возникать аномалии зубов - искривление зубного ряда, гипоплазия, склонность к кариесу. Деформации костей конечностей сводятся к расширению фаланг первых пальцев на руках и ногах, может отмечаться частичная синдактилия.
Синдром Пфайффера 2. Го типа - более тяжелая форма заболевания, проявляющаяся значительной выраженностью сращения костей черепа между собой и рядом других аномалий. Голова из-за краниосиностоза приобретает характерную форму «трилистника» или «листка клевера», наблюдается значительная гипоплазия средней трети лица. По причине грубого нарушения формирования черепа данный тип синдрома Пфайффера характеризуется выраженной умственной отсталостью и рядом неврологических нарушений. Пороки развития конечностей сводятся к расширению первых пальцев, синдактилии, анкилозу локтевых суставов. Кроме того, синдром Пфайффера 2-го типа часто сопровождается различными пороками развития внутренних органов. Все вышеперечисленные нарушения могут стать причиной летального исхода в раннем возрасте.
Синдром Пфайффера 3. Го типа во многом схож по своим проявлениям с предыдущим вариантом заболевания, но краниосиностоз при этом не приводит к формированию деформации черепа в виде «листка клевера». У больных отмечается увеличение головы в высоту, пороки развития конечностей (синдактилии, анкилозы суставов), экзофтальм, неврологические нарушения. Для этого варианта синдрома Пфайффера характерно раннее развитие зубов, нередко младенцы уже рождаются с ними (натальные зубы). Также возможны пороки внутренних органов и ранняя смерть из-за совокупности нарушений.
Ассоциированные симптомы: Деформация пальцев ног. Деформация пальцев рук.
Classification
Такое различие в клинической картине и прогнозе различных вариантов синдрома Пфайффера накладывает свой отпечаток на наследование этого заболевания. Так как 1-й тип, особенно в случае своевременного выявления и правильного лечения, характеризуется сохранением нормального уровня интеллекта, фертильности и выживаемости больных, возможна аутосомно-доминантная передача патологии потомству. Более тяжелые 2-й и 3-й типы чаще всего приводят к ранней смерти или глубокой инвалидизации больных, эти варианты синдрома Пфайффера возникают только в результате спонтанных мутаций.
Diagnostics
Своевременная диагностика синдрома Пфайффера крайне важна, так как она позволяет вовремя составить схему паллиативного лечения, тем самым избежать осложнений и значительно улучшить качество жизни больного. Но это справедливо лишь в отношении 1-го типа заболевания, так как при других вариантах патологии комплекс пороков развития настолько тяжел, что практически не оставляет шансов на сохранение интеллекта, а в дальнейшем - жизни больных. Для выявления синдрома Пфайффера используют ультразвуковые методики (в том числе и в качестве пренатальной диагностики), рентгенографию, молекулярно-генетические анализы. На профилактических УЗИ во время беременности при наличии этого заболевания у ребенка во 2-3 триместре можно выявить нарушение формирования черепа, синдактилию, пороки развития внутренних органов.
Рентгенологические методы редко используют при ранней диагностике синдрома Пфайффера у детей - в основном, производят исследование черепа, которое обнаруживает наличие краниосиностоза различной степени выраженности, гипоплазию верхнечелюстных костей, изменение формы глазниц. Также на рентгене можно определить изменение формы костей больших пальцев рук и ног. Молекулярно-генетическая диагностика производится врачом-генетиком - в этом случае при подозрении на наличие синдрома Пфайффера 1-го типа производят поиск точечной мутации 252Pro-Arg методом прямого секвенирования экзона 7а гена FGFR1. В других случаях выполняют секвенирование 7-го и 9-го экзонов гена FGFR2 - именно там возникает большинство дефектов, приводящих к развитию синдрома Пфайффера. Вспомогательную роль в диагностике заболевания могут играть методы медицинской визуализации, направленные на определение аномалий развития внутренних органов.
Рентгенологические методы редко используют при ранней диагностике синдрома Пфайффера у детей - в основном, производят исследование черепа, которое обнаруживает наличие краниосиностоза различной степени выраженности, гипоплазию верхнечелюстных костей, изменение формы глазниц. Также на рентгене можно определить изменение формы костей больших пальцев рук и ног. Молекулярно-генетическая диагностика производится врачом-генетиком - в этом случае при подозрении на наличие синдрома Пфайффера 1-го типа производят поиск точечной мутации 252Pro-Arg методом прямого секвенирования экзона 7а гена FGFR1. В других случаях выполняют секвенирование 7-го и 9-го экзонов гена FGFR2 - именно там возникает большинство дефектов, приводящих к развитию синдрома Пфайффера. Вспомогательную роль в диагностике заболевания могут играть методы медицинской визуализации, направленные на определение аномалий развития внутренних органов.
Treatment
Лечение синдрома Пфайффера только симптоматическое и паллиативное - специалисты стараются устранить повышенное внутричерепное давление, обеспечить нормальное развитие нервной системы, снизить выраженность нарушений со стороны внутренних органов. Для этого широко применяют хирургические методики - разделение пальцев, пластику черепа и ряд других. Также назначают препараты, направленные на улучшение питания нервной ткани - ноотропные средства, витамины. При своевременно начатом лечении синдрома Пфайффера 1-го типа выживаемость больных значительно увеличивается. К сожалению, объем медицинской помощи зачастую недостаточен для полноценного лечения больных синдромом Пфайффера 2-го и 3-го типов.