
ICD-10 codes
Description
Глутаровая ацидурия. Это аутосомно-рецессивное заболевание, которое возникает при нарушении метаболизма аминокислот и жирных кислот и вызывается мутациями в нескольких генах. Патология проявляется мышечной дистонией, гиперкинезом и другими неврологическими нарушениями, связанными с тяжелыми нарушениями метаболического состояния, поражением внутренних органов. Для диагностики глутаровой ацидурии необходимы биохимические и молекулярно-генетические исследования, а также компьютерная томография головного мозга. Лечение включает пожизненную диетическую терапию, левокарнитин и рибофлавин, а также жидкостную терапию для облегчения судорог.
Additional facts
Глутаровая ацидурия (ГА) - редкое заболевание, которое возникает в среднем 1 раз на 200 000 новорожденных, а в Западной Европе частота патологии намного выше - примерно 1 случай на 50 000 новорожденных. В медицинской литературе описано 400 подтвержденных случаев заболевания. Заболевание не имеет патогномоничных клинических проявлений, часто протекает под видом других неврологических или метаболических нарушений, что затрудняет своевременную диагностику и лечение.
Reasons
Заболевание относится к генетическим патологиям. В зависимости от локализации мутации гена различают 2 варианта глутарового ацидоза. При типе 1 имеется дефект в гене GCDH, который кодирует фермент глутарил-КоА-дегидрогеназу (локус 19p13.2). Из более чем 200 вариантов мутации R402W является наиболее распространенным и обнаруживается у 12-40% пациентов, проживающих в Европе. Также существуют мутации, специфичные для каждого энта и изолята.
У пациентов с патологией 2 типа обнаружено повреждение генов ETFA, ETFB, ETFDH, расположенных в локусах 15q23-q25, 19q13.3-q13.4, 4q32-q35 соответственно. Каждый из этих генов отвечает за кодирование отдельных субъединиц флавопротеина, транспортирующего электроны, нарушение которого приводит к множественному дефициту дегидрогеназ ацил-КоА жирных кислот (MADD). Риск развития патологии у ребенка, если оба родителя являются носителями мутировавших генов, составляет 25% вне зависимости от пола.
У пациентов с патологией 2 типа обнаружено повреждение генов ETFA, ETFB, ETFDH, расположенных в локусах 15q23-q25, 19q13.3-q13.4, 4q32-q35 соответственно. Каждый из этих генов отвечает за кодирование отдельных субъединиц флавопротеина, транспортирующего электроны, нарушение которого приводит к множественному дефициту дегидрогеназ ацил-КоА жирных кислот (MADD). Риск развития патологии у ребенка, если оба родителя являются носителями мутировавших генов, составляет 25% вне зависимости от пола.
Pathogenesis
Патологические изменения вызваны нарушениями метаболизма лизина, гидролизина и триптофана, вызванными дефицитом некоторых ферментов метаболического цикла. В результате блокирования биохимических реакций в организме происходит накопление токсичных метаболитов - глутаровой и 3-ОН-глутаровой кислоты. При НА 2 типа дополнительно нарушается метаболизм жирных кислот.
Заболевание в основном поражает нервную систему, что связано с избирательной нейротоксичностью глутаровой кислоты, ее производных. Также глутарат подавляет активность декарбоксилазы глутаминовой кислоты, снижает уровень ГАМК в коре головного мозга и в спинномозговой жидкости. У пациентов часто поражаются печень, сердечная мышца и почки.
Заболевание в основном поражает нервную систему, что связано с избирательной нейротоксичностью глутаровой кислоты, ее производных. Также глутарат подавляет активность декарбоксилазы глутаминовой кислоты, снижает уровень ГАМК в коре головного мозга и в спинномозговой жидкости. У пациентов часто поражаются печень, сердечная мышца и почки.
Symptoms
Клинические призраки появляются в раннем детстве (до 3 лет), пик проявления приходится на период 6-18 месяцев. Обычно развернутая картина возникает под воздействием триггеров: респираторных инфекций, травм, операций. По течению заболевание делится на два варианта: острое («энцефалитоподобное»), на которое приходится до 75% случаев, и подострое (доброкачественное), на которое приходится оставшиеся 25%.
Острый вариант АГ начинается с макроцефалии, которая возникает в первые месяцы жизни. Затем под действием провоцирующих факторов состояние внезапно ухудшается: открывается неукротимая рвота, диарея, начинаются судорожные припадки. Мышечный тонус резко снижен, наблюдается скованность мышц. Как правило, бывает угнетение сознания до ступора и комы.
Легкий вариант глутаровой ацидурии проявляется задержкой психомоторного развития у детей первого года жизни. В дальнейшем ребенок теряет приобретенные ранее навыки, у него возникают серьезные нарушения ходьбы, письма, речи из-за дистонической гиперкинезии. Кроме того, у пациента могут быть эпизоды обильного потоотделения, длительная лихорадка неизвестного происхождения.
Он выпускается в 3 клинических формах. При неонатальной форме при врожденных дефектах симптомы появляются с рождения. У младенцев есть множественные лицевые дисморфизмы (гипертелоризм, гипоплазия средней линии, низкие уши), дефекты передней брюшной стенки, а у мальчиков - гипоспадия. В течение нескольких дней метаболический ацидоз усиливается, что сопровождается мышечной гипотензией, гепатомегалией и нефромгалией.
Второй вариант АГ II типа - неонатальная форма без врожденных пороков развития. Проявляется в неонатальном периоде, характеризуется тяжелым ацидозом, некетотической гипогликемией и по симптоматике напоминает предыдущую форму заболевания. Отличительной особенностью является вовлечение в процесс миокарда, что проявляется тяжелой кардиомиопатией, аритмией, сердечной недостаточностью.
Кроме того, при глутаровой ацидурии II возможна форма с поздним началом, которая называется этилмалоновой / адипиновой ацидемией. Клинические симптомы возникают в дошкольном или школьном возрасте, очень редко у взрослых. Для него характерны приступы рвоты, гипогликемии, метаболического ацидоза. Больные страдают сильными мышечными болями, мышечной слабостью, миопатическим синдромом.
Ассоциированные симптомы: Диарея у ребенка. Нарушение походки. Рвота. Судороги. Судороги в ногах.
Острый вариант АГ начинается с макроцефалии, которая возникает в первые месяцы жизни. Затем под действием провоцирующих факторов состояние внезапно ухудшается: открывается неукротимая рвота, диарея, начинаются судорожные припадки. Мышечный тонус резко снижен, наблюдается скованность мышц. Как правило, бывает угнетение сознания до ступора и комы.
Легкий вариант глутаровой ацидурии проявляется задержкой психомоторного развития у детей первого года жизни. В дальнейшем ребенок теряет приобретенные ранее навыки, у него возникают серьезные нарушения ходьбы, письма, речи из-за дистонической гиперкинезии. Кроме того, у пациента могут быть эпизоды обильного потоотделения, длительная лихорадка неизвестного происхождения.
Он выпускается в 3 клинических формах. При неонатальной форме при врожденных дефектах симптомы появляются с рождения. У младенцев есть множественные лицевые дисморфизмы (гипертелоризм, гипоплазия средней линии, низкие уши), дефекты передней брюшной стенки, а у мальчиков - гипоспадия. В течение нескольких дней метаболический ацидоз усиливается, что сопровождается мышечной гипотензией, гепатомегалией и нефромгалией.
Второй вариант АГ II типа - неонатальная форма без врожденных пороков развития. Проявляется в неонатальном периоде, характеризуется тяжелым ацидозом, некетотической гипогликемией и по симптоматике напоминает предыдущую форму заболевания. Отличительной особенностью является вовлечение в процесс миокарда, что проявляется тяжелой кардиомиопатией, аритмией, сердечной недостаточностью.
Кроме того, при глутаровой ацидурии II возможна форма с поздним началом, которая называется этилмалоновой / адипиновой ацидемией. Клинические симптомы возникают в дошкольном или школьном возрасте, очень редко у взрослых. Для него характерны приступы рвоты, гипогликемии, метаболического ацидоза. Больные страдают сильными мышечными болями, мышечной слабостью, миопатическим синдромом.
Ассоциированные симптомы: Диарея у ребенка. Нарушение походки. Рвота. Судороги. Судороги в ногах.
Possible complications
Энцефалитоподобные кризы приводят к поражению базальных ганглиев головного мозга, поэтому после их купирования пациентов беспокоят различные гиперкинезы, мышечная дистония. Чем больше приступов страдает пациент, тем тяжелее становится неврологический дефицит. В первый день кризиса возможен отек мозга и смерть. Долгосрочные последствия глутарового ацидоза включают синдром хронической аспирации, недоедание и подвывих суставов.
В 10-30% случаев на первом году жизни наблюдаются субдуральные кровоизлияния, напоминающие синдром «тряски младенца», требующий дифференциальной диагностики. Часто наблюдаются офтальмологические осложнения: офтальмопарез, кровоизлияния в сетчатку, косоглазие. Нелеченные пациенты умирают в первое десятилетие жизни из-за метаболического кризиса или синдрома Рея.
В 10-30% случаев на первом году жизни наблюдаются субдуральные кровоизлияния, напоминающие синдром «тряски младенца», требующий дифференциальной диагностики. Часто наблюдаются офтальмологические осложнения: офтальмопарез, кровоизлияния в сетчатку, косоглазие. Нелеченные пациенты умирают в первое десятилетие жизни из-за метаболического кризиса или синдрома Рея.
Diagnostics
Пациенту с подозрением на глутаровую ацидурию показано обследование у педиатра, невролога или генетика. При обследовании учитываются типичные клинические признаки заболевания, время и порядок их появления, а также семейный анамнез. Для подтверждения ГА требуется расширенная диагностика с использованием различных лабораторных и инструментальных методов, важнейшими из которых являются:
• Биохимический анализ. В крови и моче наблюдается повышенное содержание органических кислот, ацилкарнитина, при этом показатель глутаровой кислоты превышает норму в 10 и более раз. Для уточнения диагноза определяется активность глутарил-КоА дегидрогеназы в лейкоцитах и фибробластах кожи.
• ДНК-диагностика. Молекулярно-генетические исследования для определения варианта мутации путем секвенирования экзонов, флуоресцентной гибридизации проводят в специализированных центрах. В зависимости от показаний такой анализ проводят пренатально с биопсией хориона.
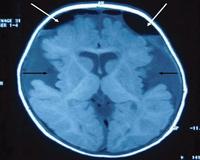
• КТ головного мозга. Характерные признаки - лобно-теменная гипоплазия, вентрикуломегалия, субдуральные гематомы. Также наблюдаются множественные очаги демиелинизации, некроза базальных ганглиев. При НА тип 1 обнаружено расширение сильвиевых трещин в виде «крыльев летучей мыши».
• Биохимический анализ. В крови и моче наблюдается повышенное содержание органических кислот, ацилкарнитина, при этом показатель глутаровой кислоты превышает норму в 10 и более раз. Для уточнения диагноза определяется активность глутарил-КоА дегидрогеназы в лейкоцитах и фибробластах кожи.
• ДНК-диагностика. Молекулярно-генетические исследования для определения варианта мутации путем секвенирования экзонов, флуоресцентной гибридизации проводят в специализированных центрах. В зависимости от показаний такой анализ проводят пренатально с биопсией хориона.
• КТ головного мозга. Характерные признаки - лобно-теменная гипоплазия, вентрикуломегалия, субдуральные гематомы. Также наблюдаются множественные очаги демиелинизации, некроза базальных ганглиев. При НА тип 1 обнаружено расширение сильвиевых трещин в виде «крыльев летучей мыши».
Treatment
Лечение следует начинать как можно скорее, при получении положительных результатов биохимического анализа, не дожидаясь ДНК-диагностики. Людям, страдающим глутаровым ацидозом, назначают специальную диету с ограничением триптофана и лизина (мясо, рыба, молочные продукты). Коррекция белкового обмена проводится с помощью лечебных белковых смесей. Медикаментозная терапия генетической патологии включает:
• Левокарнитин. Препарат связывает глутаровую кислоту, поддерживает ее выведение из организма в виде нетоксичного метаболита. Его принимают на всю жизнь, чтобы контролировать симптомы болезни.
• Рибофлавин. В редких случаях рекомендуется рибофлавин-чувствительная форма НА типа 1 для уменьшения неврологических осложнений заболевания. Улучшает метаболическое состояние пациента.
• Инфузионная терапия. В случае прорыва в мозг рекомендуется вводить растворы глюкозы с инсулином для поддержания энергетического обмена, восстановления кислотно-щелочного баланса и удаления избыточных токсичных метаболитов.
• Симптоматические средства. При обострениях глутаровой ацидурии могут применяться жаропонижающие, противорвотные препараты, антибиотики. Бензодиазепиновые транквилизаторы используются для контроля припадков.
При субдуральных гематомах, арахноидальных кистах нужна помощь детских нейрохирургов. Вопрос об удалении объемных образований головного мозга решается индивидуально с учетом их размера, локализации и степени нарушения неврологических функций. При ГА 2 типа с врожденными пороками на первом-втором году жизни их корректируют хирургическим путем и проводят пластическую операцию на лице.
• Левокарнитин. Препарат связывает глутаровую кислоту, поддерживает ее выведение из организма в виде нетоксичного метаболита. Его принимают на всю жизнь, чтобы контролировать симптомы болезни.
• Рибофлавин. В редких случаях рекомендуется рибофлавин-чувствительная форма НА типа 1 для уменьшения неврологических осложнений заболевания. Улучшает метаболическое состояние пациента.
• Инфузионная терапия. В случае прорыва в мозг рекомендуется вводить растворы глюкозы с инсулином для поддержания энергетического обмена, восстановления кислотно-щелочного баланса и удаления избыточных токсичных метаболитов.
• Симптоматические средства. При обострениях глутаровой ацидурии могут применяться жаропонижающие, противорвотные препараты, антибиотики. Бензодиазепиновые транквилизаторы используются для контроля припадков.
При субдуральных гематомах, арахноидальных кистах нужна помощь детских нейрохирургов. Вопрос об удалении объемных образований головного мозга решается индивидуально с учетом их размера, локализации и степени нарушения неврологических функций. При ГА 2 типа с врожденными пороками на первом-втором году жизни их корректируют хирургическим путем и проводят пластическую операцию на лице.
Forecast
Продолжительность жизни пациентов зависит от своевременности постановки диагноза и соблюдения врачебных предписаний. Несмотря на комплексную терапию, прогноз глутаровой ацидурии остается неблагоприятным, сохраняется высокий уровень детской смертности. Профилактика заболевания включает генетическое консультирование семей с отягощенной наследственностью и пренатальную диагностику подозреваемых случаев.
References
1. Редкий случай глутаровой ацидурии I типа у ребенка раннего возраста/ А.А. Лебеденко, С.Б. Бережанская, А.С. Тодорова, Н.Н. Вострых// Медицинский вестник Юга России. 2020. №11.
2. Глутаровая ацидурия тип I у детей. Клинические рекомендации (утв. Минздравом России). 2016.
3. Федеральные клинические рекомендации по диагностике и лечению глутаровой ацидурии тип 1. 2013.
4. Глутаровая ацидурия тип 1: клиника, диагностика и лечение/ С.В. Михайлова// Журнал неврологии и психиатрии им С.С. Корсакова. 2007. №10.
2. Глутаровая ацидурия тип I у детей. Клинические рекомендации (утв. Минздравом России). 2016.
3. Федеральные клинические рекомендации по диагностике и лечению глутаровой ацидурии тип 1. 2013.
4. Глутаровая ацидурия тип 1: клиника, диагностика и лечение/ С.В. Михайлова// Журнал неврологии и психиатрии им С.С. Корсакова. 2007. №10.