Другие названия и синонимы
Campomelic dysplasia.
МКБ-10 коды
|
|
Описание
Кампомелическая дисплазия. Летальное генетическое заболевание из группы остеохондродисплазий, характеризующееся тяжелыми скелетными аномалиями и другими нарушениями, которые нередко приводят к внутриутробной гибели плода или смерти новорожденного в первые недели жизни. Симптомами этого состояния являются сильное искривление трубчатых костей конечностей, наличие 11-ти пар ребер и колоколообразная деформация грудной клетки. В ряде случаев происходит инверсия пола. Диагностика кампомелической дисплазии чаще всего производится на основании данных пренатальных ультразвуковых исследований, кариотипирования и молекулярно-генетических исследований, после рождения ребенка для подтверждения могут использоваться данные рентгенографии. Лечения этого генетического заболевания не существует.
Дополнительные факты
Кампомелическая дисплазия (кампомелическая карликовость) - генетическое заболевание, сопровождающееся многочисленными аномалиями скелета и нарушением половой дифференцировки. Впервые патологию описал известный французский педиатр Пьер Марото в 1971-м году, однако долгое время этиология и патогенез данного состояния оставались неизвестными. Лишь в последние годы методами современной генетики удалось идентифицировать ген, дефекты которого приводят к развитию кампомелической дисплазии, и определить последовательность патологических процессов при этом состоянии. Встречаемость патологии составляет примерно 1 на 150-200 тысяч новорожденных, однако некоторые исследователи оспаривают эти цифры, указывая на повышенный риск пренатальной смерти, не учитываемый статистикой. Установлено, что кампомелическая дисплазия является аутосомно-доминантным состоянием, но практически всегда возникает по причине спонтанных мутаций у эмбриона или в половых клетках родителей. Имеются сообщения о нескольких пациентах с данной патологией, доживших до подросткового возраста.
Причины
На заре изучения кампомелической дисплазии многих врачей-генетиков ставило в тупик необычное сочетание нарушений - скелетных аномалий конечностей, лопаток, ребер и патологии черепа (расщелина твердого нёба, микроцефалия) с гермафродитизмом и половой инверсией. Причина столь необычного сочетания крылась в гене, мутации которого становятся причиной заболевания - гене SOX-9, расположенном на 17-й хромосоме. Ген кодирует одноименный белок, который является одним из важнейших транскрипционных факторов в организме человека. Например, он контролирует работу гена сol2A1, который локализуется на 12-й хромосоме и кодирует последовательность основной разновидности коллагена 2-го типа, принимающего участие в процессах формирования скелета и оссификации. Дефекты в структуре протеина SOX-9 ведут к резкому снижению экспрессии гена сol2A1, дефициту коллагена и как следствие - к кампомелической дисплазии.
Другой важной функцией SOX-9 является активация гена AMH, располагающегося на 19-й хромосоме. В основном этот ген экспрессируется в клетках Сертоли у мужчин в процессе эмбрионального развития. AMH кодирует так называемый мюллеров ингибирующий фактор, который приводит к разрушению Мюллерова протока, играющего важную роль в половой дифференцировке плода. При мутациях гена SOX-9 эта функция нарушается, в результате примерно 75% больных кампомелической дисплазией при кариотипе XY имеют фенотипически женские половые признаки. В прошлом из-за несовпадения гентотипа и фенотипа считалось, что данное заболевание намного чаще поражает девочек, нежели мальчиков. Многие исследователи предполагают активное участие белка SOX-9 и в контроле других генов, на что косвенно указывает наличие многих других аномалий развития при кампомелической дисплазии.
Другой важной функцией SOX-9 является активация гена AMH, располагающегося на 19-й хромосоме. В основном этот ген экспрессируется в клетках Сертоли у мужчин в процессе эмбрионального развития. AMH кодирует так называемый мюллеров ингибирующий фактор, который приводит к разрушению Мюллерова протока, играющего важную роль в половой дифференцировке плода. При мутациях гена SOX-9 эта функция нарушается, в результате примерно 75% больных кампомелической дисплазией при кариотипе XY имеют фенотипически женские половые признаки. В прошлом из-за несовпадения гентотипа и фенотипа считалось, что данное заболевание намного чаще поражает девочек, нежели мальчиков. Многие исследователи предполагают активное участие белка SOX-9 и в контроле других генов, на что косвенно указывает наличие многих других аномалий развития при кампомелической дисплазии.
Диагностика
Основную роль в определении кампомелической дисплазии играют методики пренатальной диагностики: ультразвуковое исследование, кариотипирование и молекулярно-генетические анализы. Ультразвуковыми методами данное заболевание можно выявить уже со 2-го триместра вынашивания ребенка, ведущими проявлениями будет искривление костей конечностей, наличие 11-ти пар ребер и колоколообразная форма грудной клетки. Также могут определяться пороки развития черепа, лица, сердца и почек, гипоплазия лопаток и тел позвонков, увеличение размеров тазового отверстия. На основании перечисленных данных пренатального ультразвукового исследования можно обоснованно предположить наличие кампомелической дисплазии и поставить вопрос о прерывании беременности по медицинским показаниям.
Кариотипирование производят с целью исключения анеуплоидий или хромосомных аномалий, которые могут вызывать схожие проявления и пороки развития плода. При этом в случае кампомелической дисплазии определение кариотипа может указать на мужской пол плода, тогда как по результатам ультразвуковых исследований будет определяться девочка. Это служит дополнительным подтверждением данного заболевания, поскольку именно для него характерны гермафродитизм и половые инверсии. Для наиболее точной диагностики могут использоваться молекулярно-генетические анализы методом секвенирования последовательности гена SOX-9. Обнаружение мутаций в данном гене является окончательным подтверждением наличия кампомелической дисплазии.
Кариотипирование производят с целью исключения анеуплоидий или хромосомных аномалий, которые могут вызывать схожие проявления и пороки развития плода. При этом в случае кампомелической дисплазии определение кариотипа может указать на мужской пол плода, тогда как по результатам ультразвуковых исследований будет определяться девочка. Это служит дополнительным подтверждением данного заболевания, поскольку именно для него характерны гермафродитизм и половые инверсии. Для наиболее точной диагностики могут использоваться молекулярно-генетические анализы методом секвенирования последовательности гена SOX-9. Обнаружение мутаций в данном гене является окончательным подтверждением наличия кампомелической дисплазии.
Лечение
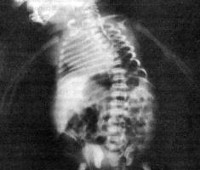
В некоторых случаях диагностику кампомелической дисплазии производят у живого новорожденного. Для этого, помимо вышеуказанных методик, применяют рентгенологическое исследование скелета. На рентгенограммах определяются многочисленные деформации трубчатых костей и другие признаки, которые были выявлены еще на УЗИ. Врачи-неонатологи также могут определить у новорожденного с кампомелической дисплазией порок сердца, аномалии развития почек и респираторные нарушения. Нередко окончательное подтверждение диагноза производится на основании данных патологоанатомического исследования. Практические любые лечебные мероприятия, даже паллиативного характера, при данном заболевании неэффективны и неспособны продлить жизнь больного.