Другие названия и синонимы
Lee Syndrome, Подострая некротизирующая энцефаломиелопатия.
МКБ-10 коды
|
|
Описание
Синдром Ли. Генетически гетерогенное наследственное заболевание, характеризующееся разнообразными нарушениями метаболизма и формирования компонентов центральной нервной системы. Симптомы этой патологии, как правило, проявляются еще в раннем детстве, к ним относят мышечную гипотонию, проблемы со вскармливанием и задержку психомоторного развития. При дальнейшем прогрессировании заболевания возникают эпилептические припадки, гиперкинезы, дыхательные расстройства. Диагностика синдрома Ли осуществляется на основании данных настоящего статуса больного, магнитно-резонансной томографии, молекулярно-генетических анализов. Специфического лечения данной патологии не существует, симптоматическая терапия лишь незначительно замедляет прогрессирование заболевания.
Дополнительные факты
Синдром Ли (подострая некротизирующая энцефаломиелопатия) - наследственное нейродегенеративное заболевание центральной нервной системы, которое характеризуется ранним началом и неуклонным прогрессированием неврологических нарушений. Впервые данное состояние было описано в 1951 году английским психиатром Денисом Ли, который определил его как наследственный вариант энцефаломиелопатии. Дальнейшие исследования показали, что синдром Ли является крайне гетерогенным состоянием с точки зрения этиологии - его причиной становятся дефекты множества генов, расположенных на аутосомах, Х-хромосоме и митохондриальной ДНК. По этой причине механизм наследования заболевания может быть (в зависимости от характера мутации) аутосомно-рецессивным, сцепленным с полом или митохондриальным. Из-за разнообразия генетических дефектов, являющихся причиной синдрома Ли, различается и половое распределение этого состояния, однако, по мнению многих врачей-генетиков, в целом можно считать, что оно в равной степени поражает как мальчиков, так и девочек. Встречаемость составляет ориентировочно 1 случай на 34-36 тысяч новорожденных.
Причины
Причинами развития синдрома Ли могут выступать мутации широкого спектра генов, расположенных на разных хромосомах. Однако патогенез этого состояния примерно сходен у различных форм патологии и чаще всего связан с нарушением процессов клеточного дыхания и функционирования дыхательной цепи митохондрий. В отношении некоторых форм синдрома Ли также замечено нарушение функционирования пируватдегидрогеназного комплекса. Нарушение структуры белков дыхательной цепи митохондрий приводит к недостаточному синтезу АТФ, являющемуся основным источником энергии во всех клетках организма. Нейроны и клетки нейроглии особенно чувствительны к недостатку энергии, что становится причиной развития разнообразных нарушений еще с детского возраста. Классификация всех генетических дефектов при синдроме Ли основана на том, какой компонент дыхательной цепи (представляющей собой белковый комплекс) митохондрий нарушен в результате мутации.
1. Синдром Ли, обусловленный поражением комплекса 1 (НАДН-KoQ-редуктаза). Этот вариант может наследоваться как аутосомно-рецессивно, так и митохондриально. Наиболее распространенные варианты заболевания этого типа обусловлены мутациями ядерных генов NDUFA10 (расположен на 19-й хромосоме), NDUFS4 и DUFAF2 (5-я хромосома), NDUFS3 (11-я хромосома). Кроме того, к развитию синдрома Ли в результате поражения митохондриального комплекса 1 способны приводить дефекты митохондриальной ДНК - генов MTND1, MTND2 и MTND3. Результатом этого является нарушение начального этапа переноса электронов и водорода в дыхательной цепи, что снижает синтез АТФ.
2. Синдром Ли, вызванный дефектами белков, входящих в митохондриальный комплекс 2 (сукцинат-KoQ-редуктаза). Заболевание этого типа наследуется только аутосомно-рецессивно, достоверно удалось установить взаимосвязь между синдромом Ли и мутациями гена SDHA, локализованного на 5-й хромосоме. Данный ген кодирует субъединицу А сукцинатдегидрогеназного комплекса, при генетических нарушениях такого типа активность фермента снижается, что и ведет к развитию заболевания.
3. Синдром Ли, возникающий в результате нарушения структуры белков митохондриального комплекса 3 (KoQН2-цитохром с-редуктаза). К этой разновидности относят наиболее распространенный вариант заболевания, вызванный мутацией гена вCS1L, расположенного на 2-й хромосоме. Развитие синдрома Ли обусловлено пониженной экспрессией фермента убихинон-с-редуктазы, входящего в состав митохондриального комплекса 3. Его выделение регулируется специфическим белком, который кодируется геном вCS1L - в результате мутации полученный дефектный протеин не способен полноценно выполнять свои функции. Для этого варианта синдрома Ли характерно аутосомно-рецессивное наследование.
4. Синдром Ли, обусловленный повреждением митохондриального комплекса 4 (цитохром с-оксидаза). Может быть вызван как мутациями ядерных генов (COX10, SCO1), в основном расположенных на 17-й хромосоме, так и повреждением митохондриальной ДНК - это удалось выяснить по характеру наследования некоторых форм, однако ключевые гены пока не определены.
5. Синдром Ли, вызванный нарушением структуры митохондриального комплекса 5 (АТФ-синтаза). К этому варианту относят сравнительно редкие мутации гена ATPAF2, локализованного на 17-й хромосоме. В результате мутации нарушается работа АТФ-синтазы, образование АТФ окислительным путем резко снижается.
1. Синдром Ли, обусловленный поражением комплекса 1 (НАДН-KoQ-редуктаза). Этот вариант может наследоваться как аутосомно-рецессивно, так и митохондриально. Наиболее распространенные варианты заболевания этого типа обусловлены мутациями ядерных генов NDUFA10 (расположен на 19-й хромосоме), NDUFS4 и DUFAF2 (5-я хромосома), NDUFS3 (11-я хромосома). Кроме того, к развитию синдрома Ли в результате поражения митохондриального комплекса 1 способны приводить дефекты митохондриальной ДНК - генов MTND1, MTND2 и MTND3. Результатом этого является нарушение начального этапа переноса электронов и водорода в дыхательной цепи, что снижает синтез АТФ.
2. Синдром Ли, вызванный дефектами белков, входящих в митохондриальный комплекс 2 (сукцинат-KoQ-редуктаза). Заболевание этого типа наследуется только аутосомно-рецессивно, достоверно удалось установить взаимосвязь между синдромом Ли и мутациями гена SDHA, локализованного на 5-й хромосоме. Данный ген кодирует субъединицу А сукцинатдегидрогеназного комплекса, при генетических нарушениях такого типа активность фермента снижается, что и ведет к развитию заболевания.
3. Синдром Ли, возникающий в результате нарушения структуры белков митохондриального комплекса 3 (KoQН2-цитохром с-редуктаза). К этой разновидности относят наиболее распространенный вариант заболевания, вызванный мутацией гена вCS1L, расположенного на 2-й хромосоме. Развитие синдрома Ли обусловлено пониженной экспрессией фермента убихинон-с-редуктазы, входящего в состав митохондриального комплекса 3. Его выделение регулируется специфическим белком, который кодируется геном вCS1L - в результате мутации полученный дефектный протеин не способен полноценно выполнять свои функции. Для этого варианта синдрома Ли характерно аутосомно-рецессивное наследование.
4. Синдром Ли, обусловленный повреждением митохондриального комплекса 4 (цитохром с-оксидаза). Может быть вызван как мутациями ядерных генов (COX10, SCO1), в основном расположенных на 17-й хромосоме, так и повреждением митохондриальной ДНК - это удалось выяснить по характеру наследования некоторых форм, однако ключевые гены пока не определены.
5. Синдром Ли, вызванный нарушением структуры митохондриального комплекса 5 (АТФ-синтаза). К этому варианту относят сравнительно редкие мутации гена ATPAF2, локализованного на 17-й хромосоме. В результате мутации нарушается работа АТФ-синтазы, образование АТФ окислительным путем резко снижается.
Классификация
В качестве отдельного варианта синдрома Ли часто указывают форму заболевания, обусловленную мутациями гена PDHA1, который расположен на Х-хромосоме. В результате наследование данного типа патологии является сцепленным с полом - болеют почти исключительно мальчики, тогда как женщины выступают носительницами патологических генов. Митохондриальный тип наследования синдрома Ли также имеет множество особенностей. Передача патологических генов происходит от матери к потомству и продолжается только по женской линии. Поскольку каждая митохондрия имеет собственную молекулу ДНК, в клетке одновременно присутствуют как «здоровые», так и «больные» органеллы, а при делении клеток (в том числе и при мейозе в процессе образования яйцеклеток) распределение больных генов оказывается неодинаковым. Женщины с относительно небольшим процентом «больных» митохондрий в клетках могут быть фенотипически здоровыми, но передавать их своему потомству. Невозможно точно предсказать, какое количество патологических митохондрий получит ребенок таких носителей, поэтому вероятность развития синдрома Ли у детей этих женщин неопределенная.
Клиническая картина
Проявления синдрома Ли обычно возникают на протяжении первого года жизни ребенка, иногда они могут регистрироваться в возрасте 2-5 лет, в редких случаях развитие заболевания начинается в подростковый период. Обычно первым проявлением патологии становится сонливость или, наоборот, повышенная возбудимость ребенка, у грудных детей наблюдается нарушение питания, недобор массы тела. В дальнейшем синдром Ли приводит к задержке психофизического развития, а у детей старшего возраста - к постепенной утрате уже обретенных навыков. Среди других неврологических симптомов заболевания наиболее часто отмечаются парезы, тремор конечностей, нарушение координации движения, поражение периферических нервов, снижение сухожильных рефлексов. В дальнейшем могут регистрироваться клонические судороги и эпилептические припадки.
Из-за недостатка энергии, обусловленного синдромом Ли, страдает не только нервная система, но и другие органы с высоким потреблением АТФ. В большинстве случаев у больных детей отмечается мышечная гипотония и слабость. Затрагивает заболевание и печень - орган с очень высоким потреблением энергии. У пациентов с синдромом Ли нередко выявляется увеличение печени, желтуха, иногда гепатолиенальный синдром. По мере прогрессирования патологии возникают нарушения дыхания - оно становится затрудненным, иногда приобретает характер дыхания Чейна-Стокса. У ряда больных со временем развивается миокардиодистрофия.
Ассоциированные симптомы: Судороги. Судороги в ногах. Тремор.
Из-за недостатка энергии, обусловленного синдромом Ли, страдает не только нервная система, но и другие органы с высоким потреблением АТФ. В большинстве случаев у больных детей отмечается мышечная гипотония и слабость. Затрагивает заболевание и печень - орган с очень высоким потреблением энергии. У пациентов с синдромом Ли нередко выявляется увеличение печени, желтуха, иногда гепатолиенальный синдром. По мере прогрессирования патологии возникают нарушения дыхания - оно становится затрудненным, иногда приобретает характер дыхания Чейна-Стокса. У ряда больных со временем развивается миокардиодистрофия.
Ассоциированные симптомы: Судороги. Судороги в ногах. Тремор.
|
|
Диагностика
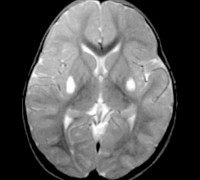
Для определения наличия синдрома Ли применяют магнитно-резонансную томографию головного мозга, электронейромиографию, изучение наследственного анамнеза, молекулярно-генетические анализы. При осмотре выявляют характерные неврологические симптомы, тремор конечностей, отставание в психофизическом развитии, у младенцев - недобор массы тела. На магнитно-резонансной томографии мозга обнаруживают симметричные изменения в области продолговатого мозга, таламуса и моста, иногда аналогичные изменения могут регистрироваться и в спинном мозге. Наилучшие результаты диагностики синдрома Ли при помощи МРТ получаются при использовании T2W и FLAIR режимов.
В тех случаях, когда имеются признаки поражения периферических нервов и мышц, для диагностики синдрома Ли выполняют электронейромиографию. При этом заболевании главным и наиболее частым результатом ЭНМР становится замедление скорости прохождения нервного импульса, которое свидетельствует о демиелинизации нервов. Изучение наследственного анамнеза информативно в случае аутосомно-рецессивных форм заболевания, при мутации генов митохондриальной ДНК четко определить семейный характер патологии затруднительно. Молекулярно-генетическая диагностика массово используется только в отношении некоторых форм синдрома Ли (обусловленных мутациями генов вCS1L, SURF1 и некоторых других).
В тех случаях, когда имеются признаки поражения периферических нервов и мышц, для диагностики синдрома Ли выполняют электронейромиографию. При этом заболевании главным и наиболее частым результатом ЭНМР становится замедление скорости прохождения нервного импульса, которое свидетельствует о демиелинизации нервов. Изучение наследственного анамнеза информативно в случае аутосомно-рецессивных форм заболевания, при мутации генов митохондриальной ДНК четко определить семейный характер патологии затруднительно. Молекулярно-генетическая диагностика массово используется только в отношении некоторых форм синдрома Ли (обусловленных мутациями генов вCS1L, SURF1 и некоторых других).
Лечение
Специфического лечения данной патологии не существует, применяется симптоматическая терапия: противосудорожные и ноотропные средства, препараты для улучшения мозгового кровообращения. Важную роль в лечении синдрома Ли играет назначение витаминов, служащих кофакторами ферментов дыхательной цепи митохондрий - В1, В6, Q10. Их регулярный прием позволяет несколько замедлить прогрессирование заболевания и уменьшить выраженность симптомов. Однако, несмотря на все предпринятые терапевтические меры, 80% больных умирает через 2-7 лет после регистрации первых проявлений патологии.