Другие названия и синонимы
Alpers disease, Синдрома Альперса-Хуттенлохера.
МКБ-10 коды
|
|
Описание
Болезнь Альперса. Это редкое митохондриальное заболевание, характеризующееся прогрессирующей энцефалопатией, связанной с циррозом печени. Патология возникает из-за мутации ДНК-гамма-полимеразы (POLG1). Синдром проявляется эпилептическими припадками, угнетением психомоторных функций, токсическим поражением печени. Для диагностики используются ЭЭГ, МРТ головного мозга, УЗИ печени, а также лабораторные исследования (биохимический анализ крови, коагулограмма, генетический тест). Поддерживающая терапия: противосудорожные препараты, метаболические препараты, парентеральное питание.
Дополнительные факты
Заболевание впервые было описано в 1931 г. Б. Альперсом у 4-месячной девочки с резистентными эпилептическими припадками. В 1976 г. P. Huttenlocher уточнил тип наследования патологии и добавил описание клинической картины. Заболеваемость синдромом Альперса-Хаттенлохера составляет 1 случай на 100 000 живорождений. Заболевание характеризуется неблагоприятным течением, быстрым ухудшением состояния больных и добавлением соматических заболеваний, поэтому, несмотря на свою редкость, не теряет своего значения в современной неврологии.
Причины
Болезнь Альперса вызывается мутацией в гене POLG1 (в локусе 15q25), который отвечает за функционирование гамма-полимеразы в митохондриях. Наиболее частыми дефектами генов являются A467T и W748S. Заболевание передается по аутосомно-рецессивному механизму, поэтому вероятность рецидива патологии при следующей беременности составляет 25%. Риск развития синдрома увеличивается при близкородственных браках, другие предрасполагающие факторы не установлены.
Патогенез
Молекулярная основа заболевания - дефицит гамма-полимеразы - единственной эукариотической ДНК-полимеразы, которая поддерживает репликацию ДНК в митохондриях. При синдроме Альперса уровень репликации генетического материала снижается до критического уровня, ухудшается активность митохондриальных ферментов. Дефицит АТФ поражает самые энергозависимые структуры - органы центральной и периферической нервной системы, печень, желудочно-кишечный тракт.
При болезни Альперса в первую очередь страдают проводники с глубоким зондированием, расположенные в задних столбах спинного мозга, а также нарушаются функции мозжечка. Вовлечение коры головного мозга в процесс происходит из-за аномальной электрической активности мозга. Приступы с лекарственной устойчивостью часто вызывают склероз гиппокампа.
При болезни Альперса в первую очередь страдают проводники с глубоким зондированием, расположенные в задних столбах спинного мозга, а также нарушаются функции мозжечка. Вовлечение коры головного мозга в процесс происходит из-за аномальной электрической активности мозга. Приступы с лекарственной устойчивостью часто вызывают склероз гиппокампа.
Клиническая картина
Типичный возраст начала заболевания составляет от 2 до 4 лет, хотя описаны случаи раннего (3-4 месяца) и позднего начала заболевания (до 8 лет). Синдром Альперса характеризуется бимодальным распределением, второй пик приходится на возраст от 17 до 24 лет. До появления первых клинических симптомов у детей нормальное психомоторное развитие и достаточный интеллект. После появления неврологических симптомов происходит резкое снижение ранее приобретенных навыков.
Болезнь Альперса в половине случаев проявляется судорогами. Это могут быть очаговые припадки, миоклонические формы (ритмические подергивания мышц без мышечных спазмов), генерализованные тонико-клонические припадки, которые именуются «большим эпилептическим пароксизмом». У детей первые приступы часто спровоцированы лихорадкой, связанной с инфекционным заболеванием.
Синдром Альперса характеризуется множественными неврологическими расстройствами. У больных наблюдается нестабильность походки, разлад движений, нарушение мелкой моторики. Сенсорная полинейропатия проявляется нарушением всех видов чувствительности, парестезиями. Как правило, возникают нарушения зрения (корковая слепота). Беспокоят мигрени, в том числе как составная часть судорожной ауры.
Важный симптом заболевания - поражение печени. Клинически это проявляется тошнотой, плохим аппетитом и нарушениями пищеварения. Часто больной ощущает слабость, сонливость, обостряется его неврологический дефицит. При подавлении синтеза факторов свертывания крови в печени открывается спонтанное кровотечение из носа, десен, легких или желудочно-кишечного тракта.
Ассоциированные симптомы: Высокая температура тела. Нарушение походки. Судороги. Судороги в ногах. Тонико-клонические судороги. Фебрильная температура тела. Шаткая походка.
Болезнь Альперса в половине случаев проявляется судорогами. Это могут быть очаговые припадки, миоклонические формы (ритмические подергивания мышц без мышечных спазмов), генерализованные тонико-клонические припадки, которые именуются «большим эпилептическим пароксизмом». У детей первые приступы часто спровоцированы лихорадкой, связанной с инфекционным заболеванием.
Синдром Альперса характеризуется множественными неврологическими расстройствами. У больных наблюдается нестабильность походки, разлад движений, нарушение мелкой моторики. Сенсорная полинейропатия проявляется нарушением всех видов чувствительности, парестезиями. Как правило, возникают нарушения зрения (корковая слепота). Беспокоят мигрени, в том числе как составная часть судорожной ауры.
Важный симптом заболевания - поражение печени. Клинически это проявляется тошнотой, плохим аппетитом и нарушениями пищеварения. Часто больной ощущает слабость, сонливость, обостряется его неврологический дефицит. При подавлении синтеза факторов свертывания крови в печени открывается спонтанное кровотечение из носа, десен, легких или желудочно-кишечного тракта.
Ассоциированные симптомы: Высокая температура тела. Нарушение походки. Судороги. Судороги в ногах. Тонико-клонические судороги. Фебрильная температура тела. Шаткая походка.
Возможные осложнения
Наиболее опасным последствием синдрома Альперса является токсический гепатит со смертельным исходом, возникающий при приеме препаратов из группы вальпроевой кислоты, снимающих судороги. При этом развивается тяжелая печеночная недостаточность, от которой пациенты умирают в течение 2-4 месяцев независимо от лечения. Повреждение гепатобилиарной системы может проявляться изменением архитектуры желчевыводящих путей, фиброзом печени.
По мере прогрессирования заболевания на фоне мышечной гипотензии развиваются дисфагия и нарушения моторики ЖКТ. Возникает необходимость введения назогастрального зонда или использования парентерального питания. Иногда поражение пищеварительной системы дополняется панкреатитом. Кардиомиопатия развивается у 10% пациентов, приводя к тяжелой сердечной недостаточности.
По мере прогрессирования заболевания на фоне мышечной гипотензии развиваются дисфагия и нарушения моторики ЖКТ. Возникает необходимость введения назогастрального зонда или использования парентерального питания. Иногда поражение пищеварительной системы дополняется панкреатитом. Кардиомиопатия развивается у 10% пациентов, приводя к тяжелой сердечной недостаточности.
Диагностика
При первичном обращении к детскому неврологу выявляется атипичная эпилептическая энцефалопатия, требующая обязательного подтверждения или исключения болезни Альперса. К обследованию пациента привлекаются генетик, гепатолог и кардиолог. Учитывая полиморфизм клинических проявлений, для верификации диагноза назначаются инструментальные и лабораторные методы исследования:
• ЭЭГ. В начале болезни определяется биполярная или гомолатеральная эпилептическая активность в виде спайков и полиспайков. Для пикового периода синдрома Альперса характерна диффузная медленная активность, преимущественно в затылочных областях с региональными акцентами в височно-теменных областях мозга.
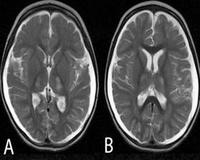
• МРТ головного мозга. На снимках видны зоны умеренного гиперинтенсивного сигнала в затылочных областях коры головного мозга. Для стойких припадков характерно появление участков повышенной интенсивности в области таламуса, продолговатого мозга. В базальных ганглиях, стволе мозга и мозжечке визуализируется прогрессирующая атрофия.
• УЗИ брюшной полости. При токсическом гепатите наблюдается умеренное увеличение печени, снижение эхогенности ее паренхимы. Для выяснения характера патологии проводится биопсия органа, по результатам которой выявляется жировая инфильтрация гепатоцитов, разрастание желчевыводящих путей и, при длительном существовании заболевания, фиброз печени.
• Кровавые анализы. В биохимическом анализе крови определяется гипогликемия, повышение уровня трансаминаз. На развитие печеночной недостаточности указывает гипербилирубинемия, гипераммонемия, гипоальбуминемия. При оценке коагулограммы протромбиновый индекс снижен.
• Генетический анализ. При подозрении на синдром Альперса проводится полный анализ гена POLG1 с использованием автоматического секвенирования. Верификация диагноза возможна после обнаружения одной из 60 известных на сегодняшний день мутаций. Иногда тест дополняется измерением количества копий митохондриальной ДНК.
• ЭЭГ. В начале болезни определяется биполярная или гомолатеральная эпилептическая активность в виде спайков и полиспайков. Для пикового периода синдрома Альперса характерна диффузная медленная активность, преимущественно в затылочных областях с региональными акцентами в височно-теменных областях мозга.
• МРТ головного мозга. На снимках видны зоны умеренного гиперинтенсивного сигнала в затылочных областях коры головного мозга. Для стойких припадков характерно появление участков повышенной интенсивности в области таламуса, продолговатого мозга. В базальных ганглиях, стволе мозга и мозжечке визуализируется прогрессирующая атрофия.
• УЗИ брюшной полости. При токсическом гепатите наблюдается умеренное увеличение печени, снижение эхогенности ее паренхимы. Для выяснения характера патологии проводится биопсия органа, по результатам которой выявляется жировая инфильтрация гепатоцитов, разрастание желчевыводящих путей и, при длительном существовании заболевания, фиброз печени.
• Кровавые анализы. В биохимическом анализе крови определяется гипогликемия, повышение уровня трансаминаз. На развитие печеночной недостаточности указывает гипербилирубинемия, гипераммонемия, гипоальбуминемия. При оценке коагулограммы протромбиновый индекс снижен.
• Генетический анализ. При подозрении на синдром Альперса проводится полный анализ гена POLG1 с использованием автоматического секвенирования. Верификация диагноза возможна после обнаружения одной из 60 известных на сегодняшний день мутаций. Иногда тест дополняется измерением количества копий митохондриальной ДНК.
|
|
Лечение
Сложность лечения людей с болезнью Альперса связана с отсутствием эффективных этиопатогенетических препаратов. Лечащий врач индивидуально подбирает пациенту симптоматическое лечение, которое устраняет неврологические нарушения, стабилизирует параметры гомеостаза. Лечение проводится в неврологических отделениях или отделениях реанимации, используются следующие группы препаратов:
• Антиоксиданты из группы витаминоподобных веществ. Они обладают положительным анаболическим действием, улучшают параметры обмена веществ. Кроме того, это единственный способ остановить печеночную недостаточность, вызванную вальпроатом.
• Противосудорожные препараты. Для устранения судорог рекомендуется использование современных противосудорожных средств из группы моносахаридов и сульфатзамещенных ламотриджинов, не вызывающих токсический гепатит. Если они неэффективны, используются препараты старшего поколения (барбитураты, бензодиазепины).
• Растворы для инфузий. Для коррекции водно-электролитного обмена назначают растворы кристаллоидов, по показаниям вводят препараты глюкозы, смеси аминокислот для поддержания адекватного уровня энергетического обмена.
Поскольку заболевание всегда заканчивается смертью пациента, необходимо оказать комплексную помощь и медицинскую помощь по окончании болезни. Лечение включает внутривенную гидратацию и введение питательных смесей, купирование приступов, устранение гипоксии путем гипербарической оксигенации, подачу кислорода, искусственную вентиляцию легких. В рамках паллиативной помощи членам семьи пациента оказывается социально-психологическая помощь.
• Антиоксиданты из группы витаминоподобных веществ. Они обладают положительным анаболическим действием, улучшают параметры обмена веществ. Кроме того, это единственный способ остановить печеночную недостаточность, вызванную вальпроатом.
• Противосудорожные препараты. Для устранения судорог рекомендуется использование современных противосудорожных средств из группы моносахаридов и сульфатзамещенных ламотриджинов, не вызывающих токсический гепатит. Если они неэффективны, используются препараты старшего поколения (барбитураты, бензодиазепины).
• Растворы для инфузий. Для коррекции водно-электролитного обмена назначают растворы кристаллоидов, по показаниям вводят препараты глюкозы, смеси аминокислот для поддержания адекватного уровня энергетического обмена.
Поскольку заболевание всегда заканчивается смертью пациента, необходимо оказать комплексную помощь и медицинскую помощь по окончании болезни. Лечение включает внутривенную гидратацию и введение питательных смесей, купирование приступов, устранение гипоксии путем гипербарической оксигенации, подачу кислорода, искусственную вентиляцию легких. В рамках паллиативной помощи членам семьи пациента оказывается социально-психологическая помощь.
Прогноз
Болезнь Альперса имеет неблагоприятно прогрессирующее течение, смерть наступает через 3-4 года после появления клинических симптомов. Комплексная медицинская помощь облегчает состояние пациентов, но вылечить болезнь пока не удается. Основной метод первичной профилактики - медико-генетическое консультирование супружеских пар с отягощением наследственности.
Контроль показателей крови при лечении больных эпилепсией препаратами вальпроевой кислоты имеет решающее значение во вторичной профилактике заболевания. При повышении показателей функции печени и уменьшении количества тромбоцитов неврологам следует предположить возможность митохондриальной патологии и немедленно направить пациента на консультацию в генетический центр.
Контроль показателей крови при лечении больных эпилепсией препаратами вальпроевой кислоты имеет решающее значение во вторичной профилактике заболевания. При повышении показателей функции печени и уменьшении количества тромбоцитов неврологам следует предположить возможность митохондриальной патологии и немедленно направить пациента на консультацию в генетический центр.
Список литературы
1. Синдром Альперса-Хуттенлохера во врачебной практике/ М.И. Душар, Г.Р. Акопян, Л.И. Волос, О.Я. Ковалюк// Современная педиатрия. 2019. №5.
2. Синдром Альперса-Хуттенлохера/ Т.Т. Батышева, В.М. Трепилец, Л.Я. Ахадова, Г.С. Голосная// Эпилепсия и пароксизмальные состояния. 2015. №1.
3. Эпилепсия у детей с митохондриальными заболеваниями: особенности диагностики и лечения/ Н.Н. Заваденко, А.А. Холин// Эпилепсия и пароксизмальные состояния. 2012. №2.
2. Синдром Альперса-Хуттенлохера/ Т.Т. Батышева, В.М. Трепилец, Л.Я. Ахадова, Г.С. Голосная// Эпилепсия и пароксизмальные состояния. 2015. №1.
3. Эпилепсия у детей с митохондриальными заболеваниями: особенности диагностики и лечения/ Н.Н. Заваденко, А.А. Холин// Эпилепсия и пароксизмальные состояния. 2012. №2.