Другие названия и синонимы
Achondrogenesis.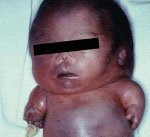
Описание
Ахондрогенез. Группа наследственных заболеваний, относящихся к классу тяжелых скелетных дисплазий, часто приводящих к антенатальной смерти или летальному исходу в раннем возрасте. Основным проявлением этих патологий является резкое нарушение процессов окостенения, которое внешне проявляется укорочением конечностей, недоразвитием ребер, грудной клетки, позвоночника. Диагностика производится на основании рентгенологических данных, а также генетических исследований - поиска мутаций в ассоциированных с ахондрогенезом генах. Специфическое лечение отсутствует, возможна только поддерживающая терапия и реанимационные мероприятия.
Дополнительные факты
Ахондрогенез - несколько генетических заболеваний, которые характеризуются недоразвитием костных и хрящевых элементов скелета. Термин «ахондрогенез» был предложен в 1952 году итальянским врачом Марко Фраккаро, в его честь названа одна из форм этой патологии. На основании рентгенологических данных в настоящее время выделяют три типа ахондрогенеза (1а, 1b, 2), которые характеризуются разным способом наследования и имеют разницу в клиническом течении. Методами генетики было выяснено, что каждой форме ахондрогенеза соответствует мутация определенного гена - указанные гены кодируют белки, напрямую участвующие в процессах окостенения и формирования хрящевой и костной тканей. Заболевание с равной долей вероятности поражает как мальчиков, так и девочек, встречаемость определена только для ахондрогенеза типа 2 и составляет примерно 1:50000 новорожденных. Механизм наследования как аутосомно-доминантный, так и аутосомно-рецессивный, однако, учитывая тяжесть ахондрогенеза и тот факт, что большинство больных не доживает до репродуктивного возраста, большую роль в развитии заболевания играют спонтанные соматические мутации. Всего достоверно описано около 100 случаев патологии.
Причины
Нарушения образования элементов скелета при ахондрогенезе обусловлены мутациями генов, которые кодируют протеины, участвующие в процессах формирования соединительных тканей. Так, причиной ахондрогенеза 1а типа (синдром Хьюстона-Харриса) являются нарушения в структуре гена TRIP1, локализованного в 17-й хромосоме - он кодирует белок 210, ассоциированный с комплексом Гольджи и бета-рецепторами к тиреоидным гормонам. Мутация такого типа наследуется по аутосомно-рецессивному механизму, то есть у здоровых родителей-гетерозигот с вероятностью 25% может родиться больной ахондрогенезом ребенок. Дефекты в структуре белка 210, обусловленные мутацией, ведут к многочисленным нарушениям формирования не только скелета, но и сердца, и легких, что наиболее часто становится причиной смерти ребенка вскоре после рождения.
Ахондрогенез 1b типа (синдром Фраккаро) вызван мутацией гена SLC26A2, локализованного на 5-й хромосоме. Он кодирует белок-переносчик сульфат-ионов, который принимает непосредственное участие в сульфонировании протеогликанов соединительных тканей, в основном хрящей. Нарушения этого процесса по причине дефекта переносчика приводят к замедлению формирования межклеточного матрикса в хрящевой ткани, а также останавливают образование эндохондральных костей, что и приводит к ахондрогенезу. В неонатологии существуют и другие наследственные дисплазии, обусловленные мутацией гена SLC26A2 - в частности, ателостеогенез. Мутации этого гена, приводящие к ахондрогенезу, наследуются по аутосомно-рецессивному механизму.
Ахондрогенез 2 типа (синдром Лангера-Салдино) является наиболее часто встречающейся патологией из этой группы. Он обусловлен мутацией гена сOL2A1, расположенного на 12-й хромосоме, он кодирует белок коллаген 2-го типа класса альфа-1. Этот протеин является структурным элементом хрящевой и костной тканей, поэтому дефекты в его структуре делают невозможным нормальное формирование скелета и ведут к ахондрогенезу. Мутации гена сOL2A1 могут наследоваться по аутосомно-доминантному механизму, но наибольшую роль в возникновении ахондрогенеза 2 типа играют спонтанные мутации de novo. Данная разновидность считается наиболее легкой формой скелетной дисплазии и по наблюдениям неонатологов характеризуется большей продолжительностью жизни, нежели при других вариантах ахондрогенеза.
Ахондрогенез 1b типа (синдром Фраккаро) вызван мутацией гена SLC26A2, локализованного на 5-й хромосоме. Он кодирует белок-переносчик сульфат-ионов, который принимает непосредственное участие в сульфонировании протеогликанов соединительных тканей, в основном хрящей. Нарушения этого процесса по причине дефекта переносчика приводят к замедлению формирования межклеточного матрикса в хрящевой ткани, а также останавливают образование эндохондральных костей, что и приводит к ахондрогенезу. В неонатологии существуют и другие наследственные дисплазии, обусловленные мутацией гена SLC26A2 - в частности, ателостеогенез. Мутации этого гена, приводящие к ахондрогенезу, наследуются по аутосомно-рецессивному механизму.
Ахондрогенез 2 типа (синдром Лангера-Салдино) является наиболее часто встречающейся патологией из этой группы. Он обусловлен мутацией гена сOL2A1, расположенного на 12-й хромосоме, он кодирует белок коллаген 2-го типа класса альфа-1. Этот протеин является структурным элементом хрящевой и костной тканей, поэтому дефекты в его структуре делают невозможным нормальное формирование скелета и ведут к ахондрогенезу. Мутации гена сOL2A1 могут наследоваться по аутосомно-доминантному механизму, но наибольшую роль в возникновении ахондрогенеза 2 типа играют спонтанные мутации de novo. Данная разновидность считается наиболее легкой формой скелетной дисплазии и по наблюдениям неонатологов характеризуется большей продолжительностью жизни, нежели при других вариантах ахондрогенеза.
Классификация
В настоящий момент выделяют три клинические формы ахондрогенеза, при этом тип 1 (синдром Паренти-Фраккаро) разделяют на две разновидности - 1а и 1b.
Ахондрогенез тип 1a. Обусловлен мутацией гена TRIP1, локализованного на 17 - й хромосоме, наследуется по аутосомно - рецессивному механизму. Это заболевание имеет такие особенности, как переломы ребер, наличие шипообразных отростков у проксимального метафиза бедренных костей и частое недоразвитие внутренних органов, главным образом сердца и легких.
Ахондрогенез тип 1b. Вызван мутацией гена SLC26A2, расположенного на 5 - й хромосоме, также наследуется по аутосомно - рецессивному принципу. При этой форме патологии целостность грудной клетки, как правило, сохранена, шипообразные отростки при рентгеновском исследовании обнаруживаются у дистальных метафизов бедренных костей. При гистологическом исследовании хрящей выявляется снижение объема межклеточного матрикса при обилии коллагеновых волокон.
Ахондрогенез тип 2. Обусловлен мутацией гена сOL2A1, расположенного на 12 - й хромосоме, механизм наследования - аутосомно - доминантный, однако большую роль играют спонтанные соматические мутации. Рентгенологическими особенностями этой формы заболевания является резкое снижение оссификации позвоночника и крестцовой кости.
Данные о выявлении 3-го и 4-го типа ахондрогенеза на сегодняшний день являются предметом научных споров. Врачи-генетики в настоящее время не подтвердили открытия мутаций новых генов, которые бы вызывали такие нарушения.
Ахондрогенез тип 1a. Обусловлен мутацией гена TRIP1, локализованного на 17 - й хромосоме, наследуется по аутосомно - рецессивному механизму. Это заболевание имеет такие особенности, как переломы ребер, наличие шипообразных отростков у проксимального метафиза бедренных костей и частое недоразвитие внутренних органов, главным образом сердца и легких.
Ахондрогенез тип 1b. Вызван мутацией гена SLC26A2, расположенного на 5 - й хромосоме, также наследуется по аутосомно - рецессивному принципу. При этой форме патологии целостность грудной клетки, как правило, сохранена, шипообразные отростки при рентгеновском исследовании обнаруживаются у дистальных метафизов бедренных костей. При гистологическом исследовании хрящей выявляется снижение объема межклеточного матрикса при обилии коллагеновых волокон.
Ахондрогенез тип 2. Обусловлен мутацией гена сOL2A1, расположенного на 12 - й хромосоме, механизм наследования - аутосомно - доминантный, однако большую роль играют спонтанные соматические мутации. Рентгенологическими особенностями этой формы заболевания является резкое снижение оссификации позвоночника и крестцовой кости.
Данные о выявлении 3-го и 4-го типа ахондрогенеза на сегодняшний день являются предметом научных споров. Врачи-генетики в настоящее время не подтвердили открытия мутаций новых генов, которые бы вызывали такие нарушения.
Диагностика
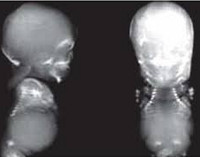
Постановка диагноза «ахондрогенез» основывается на оценке настоящего статуса пациента, рентгенологических данных, генетических исследований и изучении наследственного анамнеза и гистологического строения хрящей. На рентгенограммах костей обнаруживается укорочение длинных трубчатых костей, нарушения окостенения позвоночного столба, ребер, часто выявляют патологические переломы последних (особенно при ахондрогенезе тип 1а). Снижение оссификации также наблюдается в тазовых костях и на крестце. На метафизах бедренных костей при ахондрогенезе обнаруживаются шипообразные отростки.
В случае ахондрогенеза тип 1а при гистологическом изучении хрящей выявляется сохранение межклеточного матрикса, хондроциты имеют большое количество включений в виде вакуолей. При типе 1b напротив, наблюдается резкое снижение объема межклеточного вещества, при этом наблюдается большое количество коллагеновых волокон, которые нередко окружают хондроциты в виде плотного кокона. Гистологическая картина при ахондрогенезе типа 2 может быть довольно разнообразной - от полного отсутствия волокон в матриксе до картины, сходной с заболеванием типа 1b.
Генетическая диагностика ахондрогенеза производится путем секвенирования генов сOL2A1 и SLC26A2 с целью выявления мутаций. В отношении гена SLC26A2 возможна также диагностика носительства у здоровых гетерозигот. Изучение наследственного анамнеза оправдано только в отношении ахондрогенеза типов 1а и 1b, имеющих аутосомно-рецессивный механизм наследования. Тип 2 в подавляющем большинстве случаев возникает по причине мутации de novo, поэтому поиск аналогичных нарушений у родственников не имеет смысла. Также возможна пренатальная диагностика ахондрогенеза при помощи ультразвуковых исследований и генетического анализа - забор материала производят методом амниоцентеза.
В случае ахондрогенеза тип 1а при гистологическом изучении хрящей выявляется сохранение межклеточного матрикса, хондроциты имеют большое количество включений в виде вакуолей. При типе 1b напротив, наблюдается резкое снижение объема межклеточного вещества, при этом наблюдается большое количество коллагеновых волокон, которые нередко окружают хондроциты в виде плотного кокона. Гистологическая картина при ахондрогенезе типа 2 может быть довольно разнообразной - от полного отсутствия волокон в матриксе до картины, сходной с заболеванием типа 1b.
Генетическая диагностика ахондрогенеза производится путем секвенирования генов сOL2A1 и SLC26A2 с целью выявления мутаций. В отношении гена SLC26A2 возможна также диагностика носительства у здоровых гетерозигот. Изучение наследственного анамнеза оправдано только в отношении ахондрогенеза типов 1а и 1b, имеющих аутосомно-рецессивный механизм наследования. Тип 2 в подавляющем большинстве случаев возникает по причине мутации de novo, поэтому поиск аналогичных нарушений у родственников не имеет смысла. Также возможна пренатальная диагностика ахондрогенеза при помощи ультразвуковых исследований и генетического анализа - забор материала производят методом амниоцентеза.
Лечение
Специфического лечения ахондрогенеза в настоящий момент не существует, возможна только поддерживающая терапия. Она сводится к реанимационным мероприятиям, поддержке дыхания и кровообращения больного.
Прогноз
Прогноз крайне неблагоприятный, если ребенок родился живым, то время его жизни составляет от нескольких часов до нескольких месяцев. Смерть в большинстве случаев наступает из-за дыхательной недостаточности, вызванной аномалией строения грудной клетки и ассоциированных с ахондрогенезом пороками внутренних органов.