Другие названия и синонимы
Myotonia.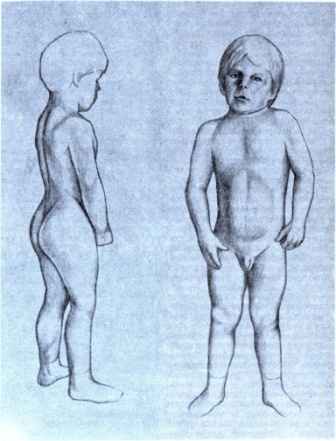
МКБ-10 коды
Описание
Миотония. Наследственное заболевание, относящееся к каналопатиям (заболевания, связанные с патологией ионных каналов). Проявляется замедленным расслаблением мышц. Характерные признаки миотонии - миотонические разряды, выявляемые игольчатой ЭМГ, и миотонические феномены, которые выявляются при клиническом обследовании. Врожденная миотония сопровождается мышечной гипертрофией, дистрофическая миотония, напротив, сопровождается мышечными атрофиями. Диагностика миотонии осуществляется при помощи ЭМГ, ЭНГ и исследования вызванных потенциалов. До настоящего времени радикальная медикаментозная терапия миотонии не разработана. Пациентам проводится симптоматическое и метаболическое лечение, массаж, ЛФК, электростимуляция.
Дополнительные факты
Миотония. Наследственное заболевание, относящееся к каналопатиям (заболевания, связанные с патологией ионных каналов). Проявляется замедленным расслаблением мышц. Характерные признаки миотонии - миотонические разряды, выявляемые игольчатой ЭМГ, и миотонические феномены, которые выявляются при клиническом обследовании.
Причины
Среди двух типов дистрофической миотонии тип 1, ген которого картирован в локусе 19q13, встречается наиболее часто (98%). Как и все типы дистрофической миотонии он передается по наследству по аутосомно-доминантному типу. Основной этиологический фактор дистрофической миотонии 1 типа - увеличением количества тринуклеотидных повторов сTG (до нескольких тысяч). Миотонин-протеинкиназа, кодируемая геном DMPK, присутствует не только в скелетных, но и миокарде, а также ЦНС. Этим и объясняются основные клинические проявления дистрофической миотонии.
Врожденная парамиотония Эйленбурга, связанная с патологией натриевых каналов, передается по аутосомно-доминантному типу. Ген SCN4A картирован в локусе 17q23. 1-q25. 3. Парамиотонические проявления развиваются в связи с повышенной возбудимостью мембраны мышечных волокон и нарушением функционирования сократительных элементов мышцы.
Врожденная парамиотония Эйленбурга, связанная с патологией натриевых каналов, передается по аутосомно-доминантному типу. Ген SCN4A картирован в локусе 17q23. 1-q25. 3. Парамиотонические проявления развиваются в связи с повышенной возбудимостью мембраны мышечных волокон и нарушением функционирования сократительных элементов мышцы.
Патогенез
В отличие от большинства нейромиотоний, являющихся приобретенными заболеваниями, идиопатическая нейромиотония (синдром Исаакса) передается по наследству и встречается наиболее часто. Эффективность введения кураре для купирования мышечных спазмов говорит о неврогенной природе нейромиотонии, однако окончательно патогенез заболевания неясен и сегодня. С выявлением повышенного титра антител к потенциал-зависимым калиевым каналам нейромиотонию стали считать аутоиммунным заболеванием. Об аутоиммунном характере заболевания говорит также наблюдаемая в ряде случаев эффективность плазмафереза.
Классификация
Наиболее распространенные формы миотонии:
• дистрофическая (двух типов).
• хондродистрофическая.
• конгенитальная аутосомно-доминантная.
• конгенитальная аутосомно-рецессивная.
• парамиотония Эйленбурга.
• нейромиотония.
• дистрофическая (двух типов).
• хондродистрофическая.
• конгенитальная аутосомно-доминантная.
• конгенитальная аутосомно-рецессивная.
• парамиотония Эйленбурга.
• нейромиотония.
Клиническая картина
Симптом «кулака» - основной клинический тест на выявление миотонии: пациент не может быстро разжать кулак, для этого ему нужно время и определенные усилия. При повторных попытках такой миотонический феномен угасает за исключением миотонии Эйленбурга, когда скованность, наоборот, усиливается с каждой повторной попыткой. Скованность также наблюдается при разжимании сжатых челюстей, быстро открыть зажмуренные глаза, быстро встать со стула. На игольчатой электромиографии выявляют один из самых характерных для миотонии феноменов - миотонические разряды, сопровождающиеся звуком «пикирующего бомбардировщика», возникающие при введении и перемещении игольчатого электрода.
Отличительной клинической особенностью врожденной миотонии является гипертрофия отдельных мышечных групп, которая создает впечатление об атлетическом телосложении пациента. В большинстве случаев мышечная сила сохранена, но иногда бывает снижена в дистальных мышцах рук. Дистрофическая миотония - мультисистемное заболевание. В большинстве случаев неврологические симптомы сочетаются с сердечной патологией (гипертрофия левого желудочка, аритмия), церебральными симптомами (гиперсомния, сниженный уровень интеллекта), эндокринными расстройствами (нарушение менструального цикла у женщин; гипогонадизм и импотенция у мужчин). Для парамиотонии типична т. н. «холодовая миотония» - возникновение мышечного спазма и пареза на холоде; такие приступы могут длиться от нескольких минут до нескольких часов. Клиническими проявлениями нейромиотоний являются мышечная скованность, спазмы (безболезненные) и постоянная мышечная активность на ЭМГ.
Ассоциированные симптомы: Судороги в ногах.
Отличительной клинической особенностью врожденной миотонии является гипертрофия отдельных мышечных групп, которая создает впечатление об атлетическом телосложении пациента. В большинстве случаев мышечная сила сохранена, но иногда бывает снижена в дистальных мышцах рук. Дистрофическая миотония - мультисистемное заболевание. В большинстве случаев неврологические симптомы сочетаются с сердечной патологией (гипертрофия левого желудочка, аритмия), церебральными симптомами (гиперсомния, сниженный уровень интеллекта), эндокринными расстройствами (нарушение менструального цикла у женщин; гипогонадизм и импотенция у мужчин). Для парамиотонии типична т. н. «холодовая миотония» - возникновение мышечного спазма и пареза на холоде; такие приступы могут длиться от нескольких минут до нескольких часов. Клиническими проявлениями нейромиотоний являются мышечная скованность, спазмы (безболезненные) и постоянная мышечная активность на ЭМГ.
Ассоциированные симптомы: Судороги в ногах.
Диагностика
Дебют дистрофической миотонии приходится, как правило, на юношеский либо взрослый возраст, степень прогрессирования заболевания зависит от генетического дефекта, поэтому тщательный сбор семейного анамнеза имеет большое значение в диагностировании дистрофической миотонии. Миотония Беккера дебютирует в 5-12 лет и характеризуется медленным течением, а миотония Томсена может дебютировать как в детском, так и в зрелом возрасте и протекает, как правило, тяжело и с осложнениями. Физикальное обследование при дистрофической миотонии выявляет атрофию мышц и снижение их силы. Для дистрофической миотонии типа 1 характерна мышечная слабость в дистальных отделах конечностей, для дистрофической миотонии типа 2 - в проксимальных.
С помощью лабораторных исследований при нейромиотонии выявляют антитела к потенциал-зависимым калиевым каналам, а дистрофическая миотония отличается незначительным повышением активности КФК в крови.
Основными инструментальными методом диагностики миотонии является игольчатая электромиография (ЭМГ), на которой определяют миотонические разряды - патогномоничный признак миотонии. Проводится исследование при помощи вызванных потенциалов и электронейрография. При парамиотонии на ЭМГ регистрируют нормальные ПДЕ и редкие миотонические разряды. Для врожденной миотонии типично сохранение параметров ДЕ в пределах нормы, для дистрофической миотонии - сочетание невропатических и миопатических черт. Для диагностики парамиотонии проводят холодовую пробу: незначительное охлаждение вызывает миотонические разряды, при дальнейшем охлаждении наступает «биоэлектрическое молчание» (исчезают как миотонические феномены, так и ПДЕ). ДНК-диагностика дистрофической миотонии основана на выявлении повышенного количества сTG-повторов в гене DMPK.
С помощью лабораторных исследований при нейромиотонии выявляют антитела к потенциал-зависимым калиевым каналам, а дистрофическая миотония отличается незначительным повышением активности КФК в крови.
Основными инструментальными методом диагностики миотонии является игольчатая электромиография (ЭМГ), на которой определяют миотонические разряды - патогномоничный признак миотонии. Проводится исследование при помощи вызванных потенциалов и электронейрография. При парамиотонии на ЭМГ регистрируют нормальные ПДЕ и редкие миотонические разряды. Для врожденной миотонии типично сохранение параметров ДЕ в пределах нормы, для дистрофической миотонии - сочетание невропатических и миопатических черт. Для диагностики парамиотонии проводят холодовую пробу: незначительное охлаждение вызывает миотонические разряды, при дальнейшем охлаждении наступает «биоэлектрическое молчание» (исчезают как миотонические феномены, так и ПДЕ). ДНК-диагностика дистрофической миотонии основана на выявлении повышенного количества сTG-повторов в гене DMPK.
Лечение
Целью лечения нейромиотонии является устранение постоянной мышечной активности и достижение возможной ремиссии, целью лечения миотонии - снижение выраженности миотонических проявлений. Немедикаментозное лечение миотонии состоит из диеты с ограничением солей калия, ЛФК, массажа, электромиостимуляции, а также предупреждения переохлаждений, так как при холоде усиливаются все миотонические реакции. Радикального медикаментозного лечения миотонии не существует, поэтому в целях уменьшении выраженности миотонических проявлений применяют фенитоин, а для снижения уровня калия - диуретики. В некоторых случаях удается достичь ремиссии с помощью иммуносупрессивной терапии: внутривенное введение иммуноглобулина человека, преднизолон, циклофосфамид.
Прогноз
Прогноз для жизни при миотонии в целом благоприятный за исключением редких случаев дистрофической миотонии типа 1, когда возможно наступление внезапной сердечной смерти по причине кардиальной патологии. Прогноз для трудоспособности пациентов с врожденными миопатиями также благоприятен (при рациональном трудоустройстве).